|
||||
|
|
ЧАСТЬ I. ОБЩАЯ ПАТОФИЗИОЛОГИЯ ЛЕКЦИЯ № 1. ПРОБЛЕМЫ ОБЩЕЙ ПАТОЛОГИИ Общая этиология и патогенез. Значение реактивности организма в патологии Этиология – учение о причинах и условиях возникновения и развития заболеваний и патологических процессов. Согласно диалектико-материалистическим представлениям, причиной любого заболевания является взаимодействие трех элементов: этиологического фактора, живого организма и условий среды, приводящее к нарушению структуры и функции живой системы. Этиологический фактор (ЭФ) – главный, ведущий, вызывающий фактор, без наличия которого не было бы заболевания (например, палочка Коха при туберкулезе). Этиологический фактор бывает простым (механическое воздействие) или комплексным (поражающие факторы ядерного взрыва), действующим длительно, в течение всего заболевания (микробы, вирусы, токсины), или только запускающим патологический процесс (тепловой фактор при ожоге). Важно отметить, что ЭФ не есть сама причина болезни, а только один из элементов причинного взаимодействия, поэтому утверждение, что туберкулезная палочка является причиной туберкулеза, является ошибочным (здесь перепутаны понятия «причина» и «причинный фактор»). Живой организм активно взаимодействует с этиологическим фактором, изменяя его и изменяясь сам в процессе взаимодействия. Организм обладает фундаментальным свойством – реактивностью, под которой понимают способность организма определенным образом реагировать на воздействие факторов среды. Реактивность является интегральной характеристикой целого организма, определяющей возможность и характер развития заболевания и претерпевающей изменения в процессе болезни. Выделяют несколько форм реактивности: 1) возрастную (отражает особенности реакции в различные возрастные периоды); 2) половую (отражает различия в реакции на воздействие на мужчин и женщин); 3) иммунологическую (отражает особенности реакции иммунной системы на антигенное воздействие); 4) групповую (например, предрасположенность к заболеваниям лиц с определенными группами крови); 5) индивидуальную и др. Можно говорить о местной (локальной) реактивности (например, особенности метаболизма канцерогена в определенной ткани) и общей реактивности, определяющей целостную реакцию организма на воздействие. Следует различать нормальную реактивность организма (когда реакция адекватна раздражителю) и патологически измененную реактивность (например, формирование повышенной чувствительности к аллергену при сенсибилизации). Одним из важнейших свойств организма является резистентность, т. е. способность противостоять воздействию патогенных факторов. Различают неспецифическую и специфическую резистентность (иммунитет). Неспецифическая резистентность обеспечивается барьерными системами, защитными белками (интерфероны, пропердин, комплемент, лизоцим, Р-лизины), фагоцитирующими клетками, интегральными сосудисто-тканевыми реакциями (воспаление), системными нейрогуморальными механизмами (общий адаптационный синдром). Системной неспецифической защитной реакцией является лихорадка. Иммунитет как способ специфической защиты внутренней среды организма от веществ и агентов, несущих признаки чужеродной генетической информации, реализуется за счет гуморальных механизмов (выработки защитных антител) и при участии специализированных клеток (Т-лимфоцитов). Реактивность и резистентность организма определяются множеством взаимодействующих местных и системных факторов и зависят от особенностей метаболизма, митотической активности клеток, стадии клеточного цикла, биоритмической организации системы, перемежающейся активности структур, нейрогуморального контроля и т. п. Третий элемент причинного взаимодействия – условия среды (внешней и внутренней), которые могут существенно модифицировать процесс взаимодействия этиологического фактора с организмом. Сюда относятся влияние климато-географических факторов (пример – «болезни жарких стран»), характер питания, сезонность, социальные факторы, стрессорные ситуации, температура, влажность, радиационный фон и др. Участие каждого из элементов причинного взаимодействия является обязательным для возникновения следствия (заболевания), однако их соотносительная роль в развитии болезни может быть различной. Абсолютизация любого из элементов причины лежит в основе ряда метафизических концепций в проблеме причинности в патологии. Так, попытка свести причину к воздействию одного фактора – суть монокаузализма, абсолютизация свойств макроорганизма ведет к конституционализму, наконец, взгляд на причину как на случайное сочетание равнозначных и равновероятных условий отражает представления сторонников кондиционализма. В основе любого заболевания лежит повреждение каких-либо структур живого организма, приводящее к нарушению его нормального функционирования. В роли повреждающих (альтерирующих) агентов могут выступать разнообразные экзо– и эндогенные факторы. В ряде случаев клеточно-тканевые изменения, возникающие при альтерации, достаточно очевидны (воспаление, некроз, дистрофия), однако иногда изменения, возникающие в биосистеме, минимальны и затрагивают организацию макромолекул (изменение третичной или четвертичной структуры белка, конформационные изменения в биомембранах и т. п.), что значительно затрудняет обнаружение первичного дефекта. Для врача в данном случае важна принципиальная методологическая посылка: если в организме выявляется какое-либо нарушение функции, то, несомненно, должна быть изменена и структура, ответственная за реализацию данной функции, т. е. являющаяся ее материальным (морфологическим) субстратом (единство структуры и функции). С другой стороны, при наличии измененной структуры глубокий анализ позволяет выявить и наличие функциональных сдвигов. Это положение легко может быть проиллюстрировано на достаточно простых (модельных) системах – структура фермента и его каталитическая активность, структура рецептора и его сродство к агонисту и т. п. Значительно сложнее проиллюстрировать это положение при переходе на уровень целого организма, поскольку здесь включается масса дублирующих, резервных, компенсаторных механизмов, позволяющих полноценно осуществлять сложную функцию (например, сохранение кровяного давления, поддержание постоянства рН и др.) при повреждении какого-либо регуляторного звена. И тем не менее, обнаружение измененной функции (симптома заболевания) является для врача ориентиром и сигналом для поиска структурной основы этой аномалии. Термином «патогенез» обозначается механизм развития заболевания, т. е. динамичный комплекс изменений, происходящих в живой системе при воздействии на нее патогенного фактора. Патогенез заболевания – диалектически противоречивый процесс, включающий в себя две противоположные тенденции: с одной стороны, это механизмы полома, повреждения, отклонения от нормы, а с другой – механизмы защиты, адаптации, компенсации и репарации. Борьба этих двух тенденций составляет основу и определяет направление развития болезни. Если преобладают механизмы повреждения, то имеет место прогрессирование патологического процесса, если верх берут саногенетические механизмы – начинается процесс выздоровления. Изучение патогенеза заболеваний является главной задачей патологической физиологии. Роль наследственных факторов в патологии человека. Хромосомные и молекулярные болезни Все наследуемые признаки человека записаны с помощью генетического кода в макромолекулярной структуре ДНК. Двойная спираль ДНК, взаимодействуя со щелочными белками (пистонами), образует сложную надмолекулярную структуру – хромосому. Каждая хромосома содержит одну непрерывную молекулу ДНК, имеет определенный генный состав и может передавать только ей присущую наследственную информацию. Хромосомный набор (кариотип) человека включает 22 пары аутосом и 2 половые – XX или ХУ – хромосомы. Несмотря на сложившуюся в процессе эволюции значительную стабильность генетического материала и наличие ДНК-репарирующих ферментов (энзимов, исправляющих ошибки репликации ДНК), под влиянием различных физических (ионизирующая радиация, ультрафиолетовые лучи), химических (алкирующие и другие соединения) и биологических (вирусы) факторов возможны изменения структуры ДНК – мутации. Учитывая наличие в геноме эукариот мигрирующих нуклеотидных последовательностей и транспозонов, под мутацией следует понимать изменение структуры ДНК, незапрограммированное в геноме и имеющее фенотипическое выражение. Мутации в половых клетках фенотипически проявляются в виде наследственного предрасположения или наследственного заболевания. Наследственная предрасположенность – это генетически обусловленная повышенная подверженность какому-либо заболеванию, которая реализуется в определенных условиях среды. О наследственной болезни говорят в том случае, когда повреждение ДНК проявляется без дополнительного воздействия факторов среды. Все наследственные болезни человека подразделяются на две группы: молекулярные болезни, имеющие в своей основе точечный дефект на нити ДНК, и хромосомные, для которых характерно более грубое повреждение генетического материала. Молекулярные болезни – обширная группа заболеваний, природа которых связана с повреждением отдельных генов. Сейчас известно более 2500 молекулярных болезней. Причиной данной патологии являются генные (точечные) мутации, т. е. изменения последовательности нуклеотидов в молекуле ДНК. Характер патологии зависит от локализации повреждения на нити ДНК. В генетическом аппарате эукариот имеются функционально различные участки: промотор – небольшая зона ДНК, узнающаяся РНК-полимеразой и факторами инициации транскрипции, регуляторные участки – энхансеры (усилители) и сайленсеры (ослабители) транскрипции, зона терминации транскрипции, а также структурные гены, определяющие последовательность аминокислот в молекуле белка. Большинство генов эукариот имеет прерывистую структуру: последовательности нуклеотидов, кодирующие белок (экзоны), чередуются с участками, не несущими информации (интроны). В ядре синтезируется про-м-РНК-копия всего гена. Далее здесь же в ядре все несмысловые участки вырезаются, а концы кодирующих последовательностей соединяются. Этот процесс назван сплайсингом. В одном и том же гене сплайсинг может протекать разными способами (альтернативный сплайсинг), т. е. под контролем одного гена в принципе могут синтезироваться несколько полипептидов с различной аминокислотной последовательностью. Реализуется сплайсинг с помощью специализированных ферментов, а также за счет аутокатализа, когда роль фермента (рибозима) выполняет сама про-м-РНК. Мутации, затрагивающие область промотора или регуляторные участки, приводят к изменению количества синтезируемого белкового продукта, но сам белок остается неизменным. Мутации структурного гена ведут к изменению первичной структуры белка. Мутация в области интрона может остаться без последствий, однако изменение сигнальной последовательности нуклеотидов на границе экзона и интрона может привести к нарушению процесса аутосплайсинга. При мутации экзонов возможны следующие патологические изменения: 1) при мутации со «сдвигом рамки» может синтезироваться белок с резко измененной структурой и нарушенной функцией; 2) мутация может превращать бессмысленный (терминаторный) триплет в смысловой – синтезируется полипептидная цепь большей длины, чем в норме; 3) мутация может превращать смысловой триплет в терминаторный – происходит синтез укороченной полипептидной цепи; 4) мутация может приводить к изменению смысла кодона, что вызовет замену аминокислоты в полипептидной цепи. Нарушение работы ферментов сплайсинга и рибозимов фенотипически проявляется также как мутация структурного гена. Важным этапом в реализации генетической программы является посттранскрипционная модификация м-РНК. К одному концу м-РНК присоединяется отрезок поли-А, состоящий из 50 – 200 идениловых нуклеотидов. Другой конец м-РНК подвергается кэпированию, т. е. соединяется с химической группировкой, содержащей метилгуанозин. Нарушение этих процессов приводит к сокращению времени жизни м-РНК, к ее быстрому разрушению нуклеазами и, следовательно, невозможности трансляции генетической информации. Вышедшая из ядра м-РНК соединяется с цитоплазматическими белками с образованием нуклеопротеидных частиц – информосом. При патологии информосом нарушается регулируемое поступление м-РНК в белоксинтезирующую систему. Таким образом, основу молекулярных болезней составляет нарушение синтеза различных белков организма. Патология может касаться структурных, транспортных, рецепторных, антигенных белков, но чаще всего страдают белки-ферменты и большинство молекулярных болезней носит характер энзимопатий. Если в результате мутации изменен активный центр фермента – нарушается его каталитическая активность и сродство к субстрату; если затронут аллостерический центр – нарушается регуляция активности фермента метаболитами и гормонами. Для диагностики наиболее распространенных энзимопатий используются простые экспресс-методы – так называемые скрининг-тесты (скрининг – просеивание). Скринирование энзимопатий основано на определении активности аномального фермента, изучении количества конечных продуктов реакции и предшественников, а также на выявлении необычных продуктов обмена в биологических жидкостях. При хромосомных болезнях и синдромах световая микроскопия позволяет выявить изменения хромосомного набора либо в виде анэуплоидий, т. е. изменения числа аутосом (болезнь Дауна, синдромы Эдвардса и Патау) или половых хромосом (синдромы Клайнфельтера, Шерешевского – Тернера, трисомии-Х), либо в виде изменения структуры хромосом (делеции, дупликации, инверсии, транслокации). Причиной анэуплоидий является нерасхождение хромосом в процессе митоза или мейоза. Замечено повышение частоты нерасхождения с увеличением возраста матери. В настоящее время описано более 100 различных хромосомных синдромов. Около 50 % всех случаев спонтанных абортов связаны с аномалиями хромосом. При этом хромосомные дефекты, унаследованные от предыдущих поколений, составляют лишь 1,3 % среди спонтанных абортов и 5,9 % среди мертворождений. Следовательно, чаще всего хромосомные аберрации являются результатом первичного нарушения гаметогенеза в родительском организме или появляются в процессе развития зародыша. Для диагностики хромосомных болезней проводят исследование хромосомного набора человека (кариотипа), а также определяют Х– и Y-половой хроматин, что позволяет обнаружить изменение числа половых хромосом в кариотипе. Важным экспресс-методом диагностики хромосомных болезней является исследование дерматоглифического фенотипа – наследственных особенностей кожного рисунка. ЛЕКЦИЯ № 2. МЕХАНИЗМЫ КАНЦЕРОГЕНЕЗА Онкологические заболевания занимают второе место как причина смертности населения в экономически развитых странах, уступая только заболеваниям сердечно-сосудистой системы. В разных регионах земного шара число больных опухолями колеблется от 65 до 360 на 100 000 населения. Опухоль – это избыточное, некоординируемое организмом, потенциально беспредельное разрастание ткани, состоящей из качественно измененных клеток, для которых характерны безудержная пролиферация, нарушение дифференцировки, морфологический, биохимический и функциональный атипизм. Опухолевый процесс – это несбалансированный тканевый рост, избыточное размножение клеток, не отвечающее потребностям ткани и организма в целом. В патологии встречаются и другие процессы, сопровождающиеся разрастанием ткани, но они существенно отличаются от истинного опухолевого роста. Так, одним из тканевых проявлений воспалительной реакции является пролиферация клеток. Но при воспалении пролиферируют клетки различного генеза: специфические клетки данной ткани, клетки соединительной ткани, сосудов, некоторые клетки крови. Рост же опухоли осуществляется за счет размножения клеток одного типа, являющихся потомками одной клетки, подвергшейся трансформации. Пролиферация клеток при воспалении не беспредельна, она регулируема, сопровождается клеточной дифференцировкой и продолжается до восполнения тканевого дефекта. В основе гиперплазии и регенерации также лежит размножение клеточных элементов одного типа, но и эта пролиферация не беспредельна, как в опухолях, и завершается созреванием клеток. Таким образом, самой существенной особенностью опухолевой ткани является беспредельная пролиферация клеток с нарушением процесса их дифференцировки. Классификация опухолей Различают доброкачественные и злокачественные опухоли. Это разделение основано на оценке внешних особенностей отдельных опухолевых клеток и опухоли в целом, их поведения, темпа и характера роста, влияния на организм. Доброкачественные опухоли растут медленно, годами, тогда как злокачественные отличаются быстрым ростом и могут заметно эволюционировать в течение нескольких месяцев или даже недель. Доброкачественные опухоли, увеличиваясь в размере, отодвигают (раздвигают) окружающие ткани, при пальпации подвижны и имеют ровную поверхность. Злокачественные опухоли обычно плотные, с бугристой поверхностью, прорастают соседние ткани, малоподвижны. Злокачественные новообразования, помимо выраженных изменений в соседних тканях, вызывают истощение организма, способны к распространению, образованию метастазов, рецидивам и без лечения завершаются летально. Современная международная классификация опухолей является гитогенетической. В соответствии с этой классификацией различают следующие виды опухолей: 1) эпителиальные опухоли без специфической локализации; 2) опухоли экзо– и эндокринных желез; 3) мезенхимальные опухоли; 4) опухоли меланинобразующей ткани; 5) опухоли нервной ткани и оболочек мозга; 6) опухоли системы крови; 7) тератомы. В клинической практике принята классификации опухолей по TNM: Т (от лат. «tumor») – характеризует распространение первичной опухоли; N (от лат. «nodulus») – отражает состояние регионарных лимфоузлов; М (от лат. «metastasis») – указывает на наличие или отсутствие метастазов. Цифры, добавляемые к каждому из символов (1, 2, 3, 4), обозначают: для T – местное распространение первичной опухоли, для N – степень поражения метастазами регионарных лимфоузлов, для М – отсутствие отдаленных метастазов (0) или их наличие (1). Терминология Название опухоли часто складывается из названия ткани, из которой она растет, с добавлением суффикса «-ома», указывающего на опухолевую природу процесса. Таковы липома – опухоль из жировой ткани, остеома – из костной ткани, ангиома – из сосудов и т. д. Биологические особенности опухолей Совокупность признаков, отличающих опухолевую ткань и составляющие ее клетки от нормальных предшественников, обозначается термином «атипизм» (нетипичность, необычность). Различают тканевый и клеточный атипизм. Тканевый атипизм. Опухоли могут возникать в любой ткани, из всех видов составляющих ее клеток, способных к активному делению, и поэтому могут локализоваться в любой части тела. Форма опухолей разнообразна. Опухоль может расти в виде узла и прощупываться в глубине ткани или выступать на поверхности. В коже и слизистых оболочках опухоль может иметь вид гриба на широком основании или полипа на тонкой ножке. Сосочковые и древовидные формы типичны для новообразований эпителия. Поверхность опухоли может быть гладкой или бугристой. Опухоли некоторых органов могут иметь специфическую форму. Размеры опухолей весьма разнообразны. Консистенция опухоли зависит от источника ее развития: новообразования из костной и хрящевой ткани отличаются высокой плотностью, опухоли из жировой ткани мягкие. Однако вне зависимости от природы опухоль – всегда более плотное образование, чем ткань, из которой она растет. Для злокачественных опухолей характерны как клеточный, так и тканевый атипизм, в то время как для доброкачественных – только тканевый. Клеточный атипизм. Необычность раковых клеток может рассматриваться в плане особенностей их структуры (морфологический атипизм), метаболических процессов (метаболический атипизм) и своеобразия поведения (функциональный атипизм). Морфологический атипизм. Морфологический атипизм прежде всего заключается в разнообразных формах, величине и необычном строении опухолевых клеток. Опухолевые клетки, как правило, имеют значительно большую, чем нормальные, величину и нетипичную для клеток данной ткани форму. Важнейшей особенностью опухолевых клеток является глубокая структурная перестройка их поверхностных и внутриклеточных мембран. Типичным для раковых клеток является обеднение цитоплазматических мембран рецепторами, воспринимающими регуляторные нейрогуморальные сигналы («рецепторное упрощение»). Изменяются антигенные свойства мембран опухолевых клеток. Наблюдается так называемое «антигенное упрощение», когда клетка теряет часть антигенов, ранее присутствовавших на ее поверхности; вместе с тем отмечается появление новых, необычных антигенов. Так, на поверхности раковых клеток резко снижается содержание органоспецифических антигенов, антигенов системы HLA, экспрессия которых на наружной клеточной мембране необходима для распознавания клетки Т-лимфоцитами. Уменьшение экспрессии антигенов системы HLA является одним из механизмов, благодаря которым опухолевые клетки способны ускользать от иммунного надзора. Выраженные морфологические изменения выявлены и в клеточных органеллах опухолевых клеток. Ядра имеют неправильную форму, наблюдается неодинаковая степень их окрашивания. В ядрах обычно обнаруживаются разнообразные хромосомные мутации. Изменения кариотипа являются одной из характеристик трансформированных клеток. В раковых клетках заметно уменьшается количество митохондрий, изменяется их структура. Строго специфичных для опухолевых клеток морфологических изменений (т. е. изменений, которые не были бы свойственны нормальным клеткам на определенных этапах развития) не обнаружено. В настоящее время отсутствует единый морфологический признак злокачественности клетки. Более того, полный набор указанных морфологических признаков не обязателен для всех опухолей. По морфологии одной клетки, как правило, нельзя установить ее опухолевую природу. Но при исследовании группы клеток можно с известной достоверностью поставить диагноз опухоли. На этом построена цитологическая диагностика опухолей. На начальных стадиях заболевания многие признаки злокачественности еще не проявляются, поэтому единственным достоверным методом установления характера опухоли является гистологическое исследование биопсийного материала. В прогностическом плане обычно обращается внимание на два момента: степень зрелости клеточных элементов опухолевой ткани и локализацию опухоли. Метаболический атипизм. Обмен веществ злокачественных клеток отличается от метаболизма нормальных клеток и ориентирован на обеспечение непрерывного роста и митотической активности. В опухолевой клетке появляются несвойственные нормальной клетке молекулярные формы ферментов (изоферменты). Изменение набора изоферментов способствует успешной конкуренции раковых клеток с окружающими их клетками за жизненно важные субстраты. Углеводный обмен. Опухолевые клетки захватывают глюкозу из притекающей крови, даже при ее низкой концентрации, когда нормальные клетки не способны к ее поглощению. В этом плане раковая клетка работает как «ловушка глюкозы». В опухолевых клетках интенсифицируются анаэробный и аэробный гликолиз, снижаются окислительное фосфорилирование и дыхание. Накопление лактата приводит к метаболическому ацидозу в клетке и в опухоли. В абсолютном большинстве нормальных клеток анаэробное расщепление глюкозы тормозится в присутствии кислорода. Это явление получило название «пастеровский эффект». Для опухолевой клетки характерно отсутствие эффекта Пастера: анаэробное расщепление глюкозы не только идет в присутствии кислорода, но и тормозит тканевое дыхание. Это так называемый обратный пастеровский эффект (эффект Кребтри). Ключевым ферментом гликолиза является гексокиназа, активность которой в нормальной клетке регулируется гормонами: инсулин – повышает активность фермента, глюкагон и другие контринсулярные гормоны – тормозят. В раковых клетках нередко присутствует особый изофермент гексокиназы, нечувствительный к гормональным влияниям. Белковый обмен. Для раковых клеток характерна анаболическая направленность метаболизма. Опухолевые клетки интенсивно извлекают из притекающей крови аминокислоты, становясь своеобразной «ловушкой азота». В то же время в опухолевых клетках в 50 раз интенсивнее, чем в нормальных, идет синтез аминокислот, при этом резко снижена активность ферментов, осуществляющих дезаминирование и переаминирование. Жировой обмен. Опухолевые клетки интенсивно поглощают из крови свободные жирные кислоты, различные липопротеиды, холестерин («ловушка жиров»), которые используются ими в качестве субстратов для построения липидов, входящих в состав цитоплазматических мембран. Обмен нуклеиновых кислот. В опухолевых клетках повышена активность ДНК– и РНК-полимераз, идет интенсивный синтез нуклеиновых кислот – активизируются репликация и транскрипция. Стимулируется синтез как хромосомной, так и митохондриальной ДНК. В раковых клетках низка активность нуклеаз. Функциональный атипизм. Структурно-метаболические особенности раковых клеток предопределяют необычность их поведения в процессе роста и в межклеточных взаимодействиях. 1. Важнейшей и принципиальной особенностью раковых клеток является их бессмертие (иммортализация). 2. Неограниченная способность к размножению сочетается у опухолевых клеток (прежде всего злокачественных опухолей) с нарушением их созревания (дифференцировки). 3. Трансформированные клетки, как правило, теряют способность выполнять функцию, присущую исходной ткани. Степень нарушения функции зависит от уровня дедифференцировки: обычно часть опухолевых клеток может сохранять свою тканеспецифическую функцию. Между опухолевыми клетками ослаблены силы межклеточного сцепления. Этому способствуют высокий отрицательный заряд (дзета-потенциал) раковых клеток, дефицит кальция в межклеточном контакте и уменьшение числа десмосом. Раковые клетки сравнительно легко отделяются друг от друга, что создает условия для метастазирования. Опухолевые клетки весьма неприхотливы в отношении требований к условиям роста. Размножающиеся раковые клетки способны внедряться (прорастать) в окружающие ткани (например, стенку сосуда) благодаря активной продукции и секреции «факторов инвазивности» – лизосомальных протеаз, гиалуронидазы и др. Это свойство злокачественных опухолей обозначается как способность к инвазивному росту. 4. В опухолевых клетках уменьшается потребность в факторах роста. Стадии опухолевого процесса Первая стадия трансформации (индукции) – процесс превращения нормальной клетки в опухолевую (раковую). Трансформация является результатом взаимодействия нормальной клетки с трансформирующим агентом (канцерогеном). Появление в организме раковой клетки не приводит с неизбежностью к развитию опухолевой болезни и гибели организма. Вторая стадия опухолевого процесса – стадия активации (промоции), суть которой заключается в размножении трансформированной клетки, образовании клона раковых клеток и опухоли. Растущая опухоль не является застывшим, стационарным образованием с неизменными свойствами. В процессе роста ее свойства постоянно изменяются: какие-то признаки теряются, какие-то возникают. Эта эволюция свойств опухоли получила название «опухолевая прогрессия». Прогрессия – это третья стадия опухолевого роста. Наконец, четвертая стадия – исход опухолевого процесса. Этиология опухолей (на примере рака молочной железы) Предшественницей раковой клетки в организме всегда является нормальная клетка какой-либо ткани. Факторы (агенты), способные вызвать превращение (трансформацию) нормальной клетки в опухолевую, называются канцерогенами. Канцерогены – это этиологические факторы опухолевого процесса. В зависимости от природы канцерогены подразделяются на физические, химические и биологические. К физическим канцерогенам относятся различные виды ионизирующей радиации (рентгеновские, ?-лучи, элементарные частицы – протоны, нейтроны, ?-, ?-частицы), а также ультрафиолетовое излучение. Чаще всего под влиянием радиации возникают лейкозы, опухоли легких, кожи и костей, а также эндокриннозависимые опухоли (молочной железы, репродуктивной системы, щитовидной железы). Введение в организм радиоактивных изотопов может вызвать развитие опухолей в различных органах, в первую очередь в тех, где накапливаются радиоактивные вещества. Имеются наблюдения, свидетельствующие о возможности развития опухолей в местах хронического термического повреждения и длительной механической травматизации тканей под влиянием инородных тел. Химические канцерогены представляют собой обширную группу различных по структуре соединений органической и неорганической природы. Они широко распространены в окружающей среде. Полагают, что 80 – 90 % всех злокачественных опухолей человека могут быть обусловлены химическими веществами. Принято различать следующие группы химических канцерогенов. 1. Полициклические ароматические углеводороды (ПАУ) – гетероциклические соединения, содержащие активные участки, способные взаимодействовать с молекулой ДНК (бензопирен, метилхолантрен и др.). ПАУ находятся в смоле и дыме (в том числе и в табачном), в выхлопных газах автомобилей, в пережаренных и копченых продуктах. 2. Ароматические амины и аминоазосоединения. Классическими представителями этой группы являются бензидиновые красители, а также анилин и его производные, используемые в лакокрасочной промышленности. Эти вещества являются примером канцерогенов резорбтивного действия. Нитросоединения (НС) используются в народном хозяйстве в качестве консервантов пищевых продуктов, при синтезе красителей, лекарств, полимерных материалов, пестицидов и др. Нитрозамины входят в группу канцерогенов «одной дозы», поскольку предполагается, что они способны вызывать опухолевую трансформацию клетки даже при однократном воздействии. Металлы и металлоиды. Канцерогенным эффектом обладают некоторые минеральные вещества – никель, хром, мышьяк, кобальт, свинец и др. В эксперименте они вызывают опухоли на месте инъекции. Некоторые вещества, используемые в качестве лекарственных средств, обладают канцерогенными свойствами. Это – фенацетин, фенобарбитал, диэтилстилбэстрол, эстрон, циклофосфамид, имуран, гидразид изопикотиновой кислоты и др. Химические канцерогены биологического происхождения. К этой группе относятся афлатоксины – канцерогены «одной дозы». Эндогенные бластомогенные вещества. К этой группе относятся канцерогены, образующиеся в самом организме в результате нарушения нормального метаболизма. Так, при нарушении метаболизма гормонов (эстрогенов, тироксина) образуются вещества, обладающие ко-канцерогенным эффектом. Доказаны бластомогенные свойства некоторых стероидов – метаболитов холестерина и желчных кислот. Механизмы канцерогенеза Разнообразие канцерогенных факторов и вытекающее из этого факта признание несомненной полиэтиологичности опухолей наводят на мысль о множественности путей возникновения этих заболеваний. Причин рака, действительно, много, но все канцерогены должны иметь общий конечный путь реализации своего эффекта – они должны каким-то образом затрагивать молекулу клеточной ДНК. До настоящего времени было предложено множество концепций, пытающихся объяснить механизмы превращения нормальной клетки в раковую. Большинство из этих теорий имеют лишь исторический интерес или входят как составная часть в принятую в настоящее время большинством патологов универсальную теорию онкогенеза – теорию онкогенов. Основные положения теории онкогенов были сформулированы в начале 70-х годов XX в. R. Huebner и G. Todaro, которые высказали предположение, что в генетическом аппарате каждой нормальной клетки содержатся гены, при несвоевременной активации или нарушении функции которых нормальная клетка может превратиться в раковую. Эти гены получили название «протоонкогены». Протоонкогены – это обычные (нормальные) клеточные гены, контролирующие рост, размножение и дифференцировку клеток. Некоторые протоонкогены работают лишь на ранних этапах онтогенеза, другие функционируют и в дифференцированных клетках, однако работа этих генов находится под жестким контролем. В результате мутации самих протоонкогенов или стойкого изменения их активности после мутации регуляторных генов происходит превращение протоонкогена в клеточный онкоген (с-опс). Следовательно, появление онкогена связано с неадекватной (количественной, качественной или временной) экспрессией (или активацией) протоонкогена. Как известно, общее число генов в геноме человека – около 100 000. Среди них имеется около 100 истинных протоонкогенов, т. е. клеточных генов, нарушение нормальной функции которых может привести к их превращению в онкогены и к опухолевой трансформации клетки. Протоонкогены тканеспецифичны. На сегодняшний день уже выявлено более 50 протоонкогенов, объединенных в 7 основных типов. Возможны следующие причины трансформации протоонкогена в онкоген: точечная мутация, транслокация или внутрихромосомная перестройка, амплификация, активация генов-энхансеров и/или угнетение сайленсеров, трансдукция протоонкогенов вирусами, активация промотора клеточного онкогена встроившимся геномом вируса. Для фенотипического проявления дефекта протоонкогена достаточно мутации только одного его аллеля, т. е. мутация, превращающая протоонкоген в онкоген, доминантна. Превращение протоонкогена в онкоген приводит к синтезу онкобелка – в количественном или качественном отношении измененного продукта протоонкогена. Онкобелок появляется в клетке либо в увеличенном количестве, либо приобретает измененную структуру и свойства, что обеспечивает данному белку повышенную активность и нарушает его реакцию на регуляторные воздействия. По локализации в клетке различают ядерные, цитоплазматические и мембранные онкобелки. Ядерные онкобелки (например, myc, fos, myb), работая в ядре, выполняют роль индукторов и репрессоров генома. С их влиянием связан синтез раковой клеткой необычных для данной стадии онтогенеза или для данной ткани белков (эмбриональные и гетероорганные антигены, эктопические гормоны и т. п.). Цитоплазматические онкобелки (fps, mos, fms) являются протеинкиназами, осуществляющими модификацию различных клеточных белков путем фосфорилирования остатков тирозина, серина или треонина. Эти онкобелки ответственны за изменения клеточного метаболизма и приобретение фенотипа, типичного для опухолевой клетки. Онкобелки, локализованные на наружной клеточной мембране (sre, abl, ras), могут выступать в качестве рецепторов для естественных факторов роста или сами выполнять роль факторов роста, побуждающих клетку к делению даже в отсутствие внешнего стимула. Под влиянием онкобелков нарушается регуляция клеточного роста, пролиферации и дифференцировки, создаются условия для ускоренной репликации ДНК и непрерывного деления клетки. Это геиы-супрессоры опухолей или антионкогены, являющиеся функциональными антагонистами онкогенов. В настоящее время выявлено более 10 антионкогенов, функция которых состоит в предупреждении трансформации протоонкогенов в активные онкогены, сохранении постоянства генерации клеток, индукции апоптоза в случае нарушения структуры ДНК. Наиболее изученным из антионкогенов в настоящее время является ген, кодирующий белок р53 (р – «protein», 53 – молекулярная масса 53 КДа). Установлено, что р53 – это ядерный фосфопротеин, присутствующий в небольших количествах во всех клетках. Уровень р53 в нормальных клетках резко возрастает после воздействия агентов, повреждающих ДНК, например, после действия ионизирующей радиации, УФ-лучей, различных химических мутагенов, гипоксии. Антионкогенную функцию выполняют и синтезируемые клетками разных тканей полиамины – спермин и спермидин, эти вещества участвуют в регуляции клеточной пролиферации и дифференцировки, их уровень увеличивается при росте и регенерации тканей. В то же время полиамины стабилизируют хроматин и ядерные белки за счет образования комплексов с отрицательно заряженными группами белков и ДНК. Снижение уровня полиаминов приводит к индукции апоптоза. Из вышеизложенного следует, что в основе современных представлений о механизмах канцерогенеза лежит предпосылка, что злокачественная трансформация клетки возникает в результате различных генетических событий, превращающих протоонкогены в онкогены, и/или инактивирующих гены, осуществляющие отбор, уничтожение и ограничение пролиферации мутантных клеток. В развитии метастазов различают следующие этапы: 1) инвазия – проникновение раковых клеток в сосуд или смежную ткань; 2) транспорт – перенос раковых клеток кровью или лимфой; 3) имплантация – выход раковой клетки из сосуда и фиксация на «чужом поле» (при отсутствии следующей фазы образуются «дремлющие» метастазы); 4) активация – размножение опухолевых клеток с формированием вторичного очага опухолевого роста (метастаза). Существуют три пути метастазирования: 1) лимфогенный – по лимфатическим сосудам; 2) гематогенный – по кровеносным сосудам; 3) тканевый – по межтканевым пространствам от одной из соприкасающихся тканей к другой. Так, к примеру, при раке молочной железы наиболее часто метастазирование происходит по лимфатическим путям в регионарные лимфатические узлы. Место метастазирования может зависеть от особенностей кровоснабжения и архитектоники сосудистого русла органа. Важным фактором, определяющим возможность роста опухоли на «чужом поле», является ее неоваскуляризация. Опухоль, диаметр которой превышает 2 – 4 мм, нуждается в формировании новых капиллярных сосудов, так как ее питание уже не может обеспечиваться только за счет диффузии. Опухолевые клетки способны продуцировать факторы, стимулирующие ангиогенез. Эти вещества обеспечивают врастание сосудов в опухолевый очаг путем миграции в него эндотелиальных клеток, выстилающих предсуществующие мелкие венулы из прилегающей соединительной ткани, и их размножение. Факторы, способствующие канцерогенезу Выделяют следующие факторы, способствующие канцерогенезу. I. Наследственная предрасположенность. Наличие семейных форм рака, когда среди членов одной семьи в нескольких поколениях выявляется рак одной и той же локализации. Так, наличие у матери рака молочной железы повышает риск обнаружения рака этой локализации у пробанда в 5 раз, а наличие у матери и сестры – в 10 – 15 раз. В большинстве случаев наследственная предрасположенность к раку у человека органоспецифична и передается полигенно. II. Иммунодепрессия. Защита организма от растущей опухоли обеспечивается механизмами клеточного и – в меньшей степени – гуморального иммунитета. Иммунная система распознает раковые клетки, вызывает их разрушение либо сдерживает размножение, ингибируя фазу промоции. Моноциты и макрофаги осуществляют специфический киллинг раковых клеток после их распознавания Т-лимфоцитами. К-клетки (нулевые лимфоциты и особые клетки моноцитарного ряда) уничтожают опухолевые клетки, нагруженные цитотоксическими антителами (IgM). Любая иммунодепрессия способствует опухолевому росту. Иммунодефицитные состояния различного генеза (особенно с дефектом Т-системы) предрасполагают к возникновению опухолей. Так, наиболее часто наблюдается развитие рака молочной железы на фоне снижения и клеточного, и гуморального звеньев иммунной защиты. III. Определенный эндокринный фон. В процессе канцерогенеза важную роль играют гормоны, способные стимулировать рост клеток. Это – соматолиберин и СТГ, пролактолиберин и пролактин, тиролиберин и ТТГ, меланолиберин и меланотропный гормон, гонадолиберины, эстрогены. Избыток этих гормонов, как и нарушение баланса между ними, создает условия, способствующие развитию опухолей. Примером могут служить рак молочной железы, возникающий на фоне избытка эстрогенов, рак щитовидной железы при избытке ТТГ и т. п. IV. Хронические воспалительные и вялотекущие пролиферативные процессы. При названных патологических состояниях создается благоприятный фон для действия канцерогенных факторов. V. Пожилой возраст. Опухоли – это заболевания в основном пожилых людей. Если принять во внимание, что развитие опухоли – это многостадийный процесс возникновения, накопления и реализации генетических изменений и отбора измененных клеток, становится понятным, что с возрастом повышается вероятность «накопить» необходимое количество мутаций. Влияние опухоли на организм Растущая злокачественная опухоль оказывает влияние как на непосредственно окружающие ее ткани, так и на весь организм больного. Важнейшими проявлениями системного действия опухоли являются следующие. 1. Раковая кахексия – общее истощение организма. Раковая кахексия является результатом действия множества факторов. Опухолевые клетки успешно конкурируют с нормальными за ряд витаминов и микроэлементов. Организм реагирует на растущую опухоль как на стрессорный фактор увеличением продукции глюкокортикоидов, стимулирующих катаболизм тканевых белков. 2. Иммунодепрессия. Рост злокачественной опухоли сопровождается развитием вторичного иммунодефицита, что связано, с одной стороны, с избыточной продукцией глюкокортикоидов, а с другой – с продукцией опухолью особых факторов, ингибирующих иммунный ответ хозяина и способствующих размножению трансформированных клеток. 3. Анемия. По мере развития опухолевого процесса у больных обнаруживается прогрессирующая анемия. Анемия при раковых заболеваниях имеет сложный генез. Во-первых, опухоль выделяет вещества, снижающие содержание железа в крови, угнетающие эритропоэз в костном мозге и уменьшающие продолжительность жизни эритроцитов. Во-вторых, анемия может быть результатом скрытого кровотечения вследствие прорастания опухолью стенки сосуда. В-третьих, может сказываться возникающий в организме опухоленосителя дефицит витамина B12 (фолиевой кислоты). Наконец, возможны метастазы опухоли в костный мозг. 4. Тромбозы и геморрагические осложнения. Типичным для злокачественных опухолевых процессов является развитие изменений в системе регуляции агрегатного состояния крови с развитием ДВС-синдрома. 5. Универсальное мембраноповреждающее действие. Развивается вследствие активации процессов перекисного окисления липидов. Опухоль является ловушкой витамина Е, одного из наиболее мощных естественных антиоксидантов. В клетках организма-опухоленосителя снижается активность ферментов антиоксидантной защиты – каталазы, СОД и глутатион-пероксидазы. 6. Продукция эктопических гормонов. Вследствие дерепрессии определенных локусов генома опухолевая клетка может вырабатывать несвойственные данной ткани гормоны (например, клетки рака легкого могут продуцировать АКТГ). 7. Интоксикация. Поскольку пролиферация эндотелиальных клеток и связанное с этим новообразование сосудов, как правило, отстают от роста самой опухоли, в ее центре почти всегда обнаруживаются участки некротического распада. Продукты распада опухоли могут поступать в кровь и вызывать общую интоксикацию. 8. Отеки. В генезе опухолевых отеков принимают участие следующие факторы: гипопротеинемия, повышение сосудистой проницаемости, сдавление опухолью вен и лимфатических сосудов с нарушением оттока, развитие вторичного альдостеронизма, повышенная продукция АДГ. 9. Метастазирование. В результате метастазирования возможно развитие разнообразной вторичной симптоматики. Могут возникать серьезные нарушения функции отдаленных органов (например, параличи скелетной мускулатуры при метастазах в головной или спинной мозг). 10. Психоэмоциональные нарушения. Наличие онкологической патологии воспринимается человеком как сильнейший психический cтpecс. ЛЕКЦИЯ № 3. ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА. НАРУШЕНИЕ КИСЛОТНО-ОСНОВНОГО СОСТОЯНИЯ Водно-электролитные нарушения сопровождают и утяжеляют течение многих заболеваний. Все разнообразие этих расстройств может быть подразделено на следующие основные формы: гипо– и гиперэлектролитемии, гипогидратация (обезвоживание, эксикоз) и гипергидратация. Одним из частых проявлений расстройств водно-электролитного обмена являются отеки. Отек – это избыточное накопление жидкости в межклеточном пространстве вследствие нарушения обмена воды между кровью и интерстицием на уровне капилляра. Отеки могут быть местными, т. е. локализованными в ограниченном участке тела, и генерализованными, могут быть скрытыми и явными. Как правило, генерализованные отеки становятся клинически заметными, когда объем интерстициальной жидкости увеличивается на 15 %. Массивные отеки кожи и подкожной клетчатки называют анасаркой. Скопление внеклеточной жидкости в полостях тела получило название водянки. В зависимости от этиологических факторов принято различать следующие отеки: воспалительные, токсические, аллергические, сердечные, цирротические, почечные (нефритические и нефротические), голодные (кахектические), лимфатические, нейрогенные, эндокринные. Кроме того, отеки могут развиваться в результате применения некоторых гормональных лекарственных препаратов (вазопрессина, эстрогенов), при беременности, у новорожденных, когда ребенок рождается в состоянии физиологической гипергидратации (отек новорожденных). Особенно отеки выражены у недоношенных детей. Физиологическая убыль массы, наблюдающаяся в первые дни жизни, в значительной степени объясняется уменьшением чрезмерно увеличенного содержания внеклеточной жидкости в организме. Помимо физиологической гипергидратации, у новорожденных могут наблюдаться отеки, возникшие внутриутробно вследствие интранатальных заболеваний плода при тяжелых соматических и инфекционных болезнях матери. Отеки могут быть обусловлены и наследственными факторами. Именно в детском возрасте наиболее часто имеют место проявления наследственного ангионевротического отека, связанного с дефицитом ингибитора С1-эстеразы, регулирующего активность C1-фракции комплемента и калликреина, и появлением в плазме больных особого кинина, отличающегося от брадикинина и способного увеличивать проницаемость сосудов. Наследственный ангионевротический отек чаще всего возникает при физических нагрузках, стрессовых состояниях и локализуется в подкожной клетчатке и коже, во внутренних органах, в слизистой и подслизистой дыхательных путей. Развитие отеков является результатом действия ряда, как правило, взаимосвязанных патогенетических механизмов, главными из которых являются следующие. 1. Повышение гидростатического (венозного) давления внутри сосудов. Последнее может быть связано как с увеличением сопротивления к венозному оттоку при недостаточности кровообращения, сдавлении, закупорке, сужении вен, так и расширением артериол и прекапиллярных сфинктеров, приводящим к увеличению притока крови и резкому повышению внутрикапиллярного давления. Возрастание гидростатического давления ведет к увеличению площади фильтрации в артериальной и венозной частях капилляра и нарушению реабсорбции воды в сосудистое русло. 2. Снижение онкотического давления плазмы крови в сосудах вследствие гипоальбуминемии. При этом уменьшается способность белков плазмы удерживать жидкость внутри сосудов, усиливается фильтрация из артериальной части капилляра и резко ухудшается реабсорбция жидкости в венозной части капилляра. 3. Повышение проницаемости сосудистой стенки под влиянием ряда биологически активных веществ (гистамина, серотонина, кининов, простагландинов), токсических веществ (яда змей, насекомых, бактериальных токсинов, БАВ), выраженной гипоксии. Причинами повышения проницаемости сосудистой стенки могут быть также перерастяжение капилляров (например, при артериальной гиперемии), повреждение эндотелиальных клеток (при ацидозе), нарушение структуры базальной мембраны. Указанный механизм обуславливает усиление выхода жидкости и мелкодисперсных белков через поврежденную стенку капилляра в интерстициальное пространство, что приводит к повышению гидростатического и онкотического давления интерстициальной жидкости и задержке воды в тканях. 4. Повышение гидрофильности тканей вследствие гиперосмии и гиперонкии тканей. Гиперосмия и гиперонкия тканей могут возникать вследствие накопления в них электролитов, белков, осмотически активных продуктов метаболизма, в результате альтерации тканей, снижения активного транспорта ионов через клеточные мембраны при тканевой гипоксии, нарушения вымывания электролитов и метаболитов из тканей при нарушении микроциркуляции. Гиперосмия и гиперонкия усиливают ток жидкости из капилляров в ткани. 5. Нарушение оттока лимфы в результате повреждения, сдавления или обтурации лимфатических сосудов. При этом происходит накопление в интерстициальном пространстве избыточно профильтровавшейся и не подвергшейся обратному всасыванию в сосудистое русло жидкости. 6. Нарушение нервно-гормональной регуляции водно-электролитного обмена. Как известно, решающее значение в регуляции водно-электролитного обмена придается двум гормонам: альдостерону и антидиуретическому. Увеличение продукции этих гормонов или уменьшение их инактивации в печени играет существенную роль в развитии отеков. Наибольшее значение в патогенезе отеков отводится развитию вторичного гиперальдостеронизма – гиперсекреции альдостерона, вызванной рефлекторными влияниями на клубочковую зону коры надпочечников. Вторичный гиперальдостеронизм, вызывающий развитие отеков, часто является следствием неадекватной регуляции водно-электролитного обмена и может наблюдаться при различных формах патологии, сопровождающихся гиповолемией, гипонатриемией и ишемией почек, при которых имеет место возбуждение клеток юкстагломерулярного аппарата и активация ренинангиотензиновой системы. Последняя способна стимулировать повышенную секрецию альдостерона. При гиперпродукции альдостерона усиливается активная реабсорбция натрия в почечных канальцах. Развивающаяся в этих условиях гиперосмия плазмы крови стимулирует через осморецепторы супраоптические ядра гипоталамуса, в результате чего усиливается секреция антидиуретического гормона, регулирующего проницаемость дистальных канальцев и собирательных трубочек для воды. В отсутствие антидиуретического гормона эти части нефрона практически водонепроницаемы. С увеличением количества АДГ проницаемость дистальных канальцев и собирательных трубочек нефрона для вен возрастает и происходит задержка воды в организме. Таким образом, гиперсекреция альдостерона и АДГ приводит к задержке натрия и воды в тканях и развитию отека. Следует также отметить, что в ряде случаев патогенетическим фактором развития отеков является дефицит предсердного натрийуретического гормона, возникающий, в частности, при различных формах сердечной недостаточности. В зависимости от преобладающего значения одного из вышеуказанных факторов можно выделить следующие патогенетические варианты отеков: застойные (гемодинамические), онкотические, осмотические, мембраногенные, лимфогенные. Последствия отека для организма в значительной степени зависят от особенностей этиологического фактора, степени его выраженности и локализации. Отечная жидкость сдавливает окружающие ткани, вызывает расстройства трофики и функций, что может сказаться на нарушении метаболических процессов в организме в целом. У детей водно-электролитные расстройства развиваются быстро и протекают тяжело, особенно тяжело детский организм переносит обезвоживание. Чем моложе ребенок, тем легче развиваются водно-электролитные нарушения. Это обусловлено рядом особенностей водно-солевого обмена в детском организме. Во-первых, интенсивность водного обмена, внепочечных потерь воды у детей выше по сравнению с взрослыми. Так, у детей грудного возраста 52 – 76 % воды выделяется через кожу и легкие. Высокая интенсивность водного обмена определяет повышенную потребность в воде у маленьких детей, которая почти втрое превышает таковую у взрослых. Во-вторых, у детей, особенно первого года жизни, еще недостаточно сформированы механизмы, регулирующие постоянство объема и состава водных пространств организма. У них наблюдается физиологическая гипореактивность волюмо– и осморецепторов сосудов и тканей, снижена концентрационная способность почек, затруднено выведение избытка воды и осмотически активных веществ, вследствие чего легко возникают расстройства осмотического гомеостаза при малейшем нарушении водного и пищевого баланса. Так, при введении прикорма, переводе ребенка на искусственное питание, на питание с повышенной калорийностью (особенно за счет белков), ребенку требуется дополнительное количество воды. С другой стороны, у детей (особенно раннего возраста) часто наблюдаются заболевания, сопровождающиеся рвотой, поносами, высокой температурой, что является серьезным фактором, способствующим возникновению водно-электролитных расстройств. Нарушение кислотно-основного состояния Одним из необходимых условий существования организма является поддержание постоянства кислотно-основного соотношения (КОС). Нарушение КОС неизбежно влечет за собой развитие патологических изменений в организме, вплоть до его гибели. Сдвиг рН – величины, характеризующей состояние КОС – даже на 0,1 уже вызывает выраженные нарушения со стороны сердечно-сосудистой и дыхательной систем, а смещение рН плазмы крови выше 7,8 или ниже 6,8 несовместимо с жизнью. Расстройства КОС могут быть экзогенного и эндогенного происхождения. По направлению сдвига кислотно-основного баланса различают две формы нарушений КОС – ацидоз и алкалоз. Под ацидозом понимают нарушение КОС, при котором в крови и в тканях появляется абсолютный или относительный избыток кислот и повышается концентрация свободных водородных ионов. Алкалозом называется такое нарушение КОС, при котором в организме происходит абсолютное или относительное увеличение содержания оснований и понижается концентрация свободных ионов водорода. По механизмам развития различают ацидозы и алкалозы газовые и негазовые. По степени выраженности сдвига все ацидозы и алкалозы подразделяются на компенсированные и некомпенсированные. Показателями степени компенсации сдвига являются рН крови и соотношение компонентов бикарбонатного буфера (Н2СО3 и NаНСО3). При компенсированных формах расстройств КОС изменяются абсолютные количества компонентов бикарбонатного буфера, но их соотношение не нарушается, оставаясь в пределах нормы, т. е. примерно 1:20. При этом рН крови не изменяется. При декомпенсированных сдвигах имеет место изменение не только общего количества, но и соотношения компонентов бикарбонатного буфера, что влечет за собой отклонение рН от нормы. Наиболее частой формой нарушения КОС является негазовый ацидоз. Его развитие связано с избыточным накоплением в организме ионов водорода или потерей бикарбоната из внеклеточной жидкости. В зависимости от причин развития различают следующие разновидности негазового ацидоза. Метаболический ацидоз, возникающий вследствие избыточного образования нелетучих органических кислот в организме при нарушении обмена веществ, вызванных гипоксией, голоданием, эндокринной патологией и др. Выделительный ацидоз, обусловленный задержкой кислот в организме в результате недостаточности экскреторной функции почек или избыточной потерей щелочей через желудочно-кишечный тракт (гиперсаливация, продолжительная диарея, кишечные свищи) и почки (врожденная недостаточность ферментов, ответственных за реабсорбцию бикарбонатов, гипоксическое, токсическое поражение почек). Экзогенный ацидоз, связанный с избыточным поступлением в организм кислот (при отравлениях кислотами или при длительном приеме кислотосодержащих лекарственных веществ, например, салицилатов). В ряде случаев возможно развитие комбинированных форм негазового ацидоза, в частности – сочетание метаболической и выделительной форм. Негазовый ацидоз характеризуется снижением рН крови и буферных оснований в плазме крови. Последнее связано с нейтрализующим действием бикарбонатов на нелетучие кислоты. Увеличение концентрации ионов водорода стимулирует вентиляцию легких, что приводит к компенсаторному снижению напряжения СО2. В компенсацию включаются также внутриклеточные буферные механизмы. Ионы водорода переходят, в частности, в эритроциты, из которых взамен в плазму выходят ионы натрия и кальция. Окончательная компенсация осуществляется почками, которые начинают усиленно удалять избыток водородных ионов с мочой. При негазовом ацидозе могут возникать разнообразные нарушения функций организма. При умеренном снижении рН периферические сосуды, как правило, расширяются, что приводит к снижению артериального и венозного давления, уменьшению венозного возврата крови к сердцу; нарушается работа сердца. Однако при выраженном ацидозе возможно и сужение периферических сосудов. Кровоснабжение мозга в условиях негазового ацидоза резко снижается за счет сужения просвета сосудов, питающих мозг. Понижается сродство гемоглобина к кислороду, в результате чего затрудняются образование в легких оксигемоглобина и отдача гемоглобином кислорода в тканях. Развивается гироксемия и гипоксия. Значительно нарушается водно-электролитный обмен. С мочой теряется больше (чем в норме) натрия, калия, кальция. При снижении рН крови ниже 7,2 развивается коматозное состояние. Газовый ацидоз, или респираторный, развивается при увеличении концентрации СО2 в крови. Это может быть обусловлено либо вдыханием воздуха с высоким содержанием СО2, либо нарушением выделения легкими углекислого газа вследствие нарушения проходимости воздухоносных путей, обширного поражения легочной паренхимы, подавления активности дыхательного центра или в результате недостаточности кровообращения, когда в силу резкого снижения скорости кровотока замедляется выведение СО2 из крови. Газовый ацидоз может быть острым и хроническим (избыточная полнота, обструктивная форма легочной недостаточности – хронический бронхит, эмфизема легких). Важную роль в компенсации газового ацидоза играют гемоглобиновая и белковая буферные системы. Действие бикарбоната невелико, так как он не способен в полной мере к нейтрализации СО2. Снижение рН крови вследствие увеличения напряжения СО2 стимулирует выведение избытка ионов водорода с мочой и активную реабсорбцию бикарбоната в почечных канальцах. Тяжесть нарушений в организме при газовом ацидозе зависит от степени накопления углекислого газа и от присоединения метаболического ацидоза. Умеренные компенсированные ацидозы протекают без выраженных клинических симптомов. При выраженной гиперкапнии возникают расстройства, в первую очередь, со стороны сердечно-сосудистой системы. Негазовый алкалоз развивается, когда вследствие избыточной потери нелетучих кислот или в результате поступления большого количества оснований создается их избыток. Кроме того, причиной возникновения негазового алкалоза может быть избыточная продукция альдостерона (первичный гиперальдостеронизм, уменьшение объема внеклеточной жидкости) или длительное лечение этим препаратом, а также гипокалиемия, когда в обмен на ионы калия водородные ионы перемещаются из внеклеточной жидкости в клетки. Негазовый алкалоз характеризуется повышением концентрации буферных оснований в плазме, повышением значения рН. Респираторная компенсация приводит к снижению легочной вентиляции и повышению напряжения СО2. Однако такая компенсация не может быть длительной, так как накапливающаяся углекислота стимулирует дыхание. При негазовом алкалозе максимальное значение рСО2 обычно составляет 60 мм рт. ст. В процессе компенсации участвуют внутриклеточные буферные системы, которые отдают в плазму ионы водорода, связывая катионы натрия. Часть избыточных анионов бикарбоната уходит в эритроциты, обмениваясь на ионы хлора. С мочой выделяется большое количество бикарбоната и двуосновного фосфата. При негазовом алкалозе в связи с потерей через почки большого количества натрия наступает снижение осмотического давления внеклеточной жидкости и чрезмерное выведение при этом воды, в результате чего происходит обезвоживание организма. Значительная потеря калия сопровождается нарушением функции миокарда и нервной системы. При повышении рН крови уменьшается доля ионизированного кальция, что приводит к повышению нервно-мышечной возбудимости, развитию судорог. Газовый алкалоз обусловлен усиленным выведением углекислого газа из крови через легкие при гипервентиляции. Это наблюдается во время одышки, возникающей в результате поражения мозга, при гипертермии, выраженной лихорадке, тяжелой анемии. Развитие газового алкалоза возможно при дыхании разреженным воздухом на большой высоте, при гипервентиляции во время искусственного дыхания. Как и газовый ацидоз, газовый алкалоз может быть острым и хроническим. Главным нарушением при газовом алкалозе является снижение напряжения СО2 в крови. Начальная компенсаторная реакция на респираторный алкалоз заключается в выходе ионов водорода из клеток во внеклеточную жидкость, в усилении продукции молочной кислоты. Компенсаторные реакции включают также снижение почечной экскреции ионов водорода, снижение концентрации бикарбоната в плазме за счет усиленного их выведения с мочой. Состояние ацидоза у детей возникает значительно чаще, чем у взрослых. Так, компенсированный метаболический ацидоз уже наблюдается у ребенка в первые дни после рождения даже при нормальном течении беременности и родов у матери. У детей раннего возраста обмен веществ носит ацидотический характер с образованием большого количества недоокисленных продуктов; в то же время щелочной резерв у детей в возрасте до одного года меньше, чем у взрослых. У детей возможна наследственная форма лактат-ацидоза, патогенез которого связывают с энзиматическим блоком в цикле Кребса. Вследствие этого возникает резкая активация анаэробного гликолиза и накопление его конечного продукта – молочной кислоты, содержание которой может возрастать в 10 – 20 раз. У детей раннего возраста отмечается предрасположенность к развитию газового ацидоза, что связано с недостаточным структурным и функциональным формированием аппарата дыхания. Нарушения кислотно-основного баланса ротовой жидкости играют существенную роль в развитии патологических процессов в ротовой полости и прежде всего в развитии множественного кариеса. Минерализующие свойства слюны, обусловленные перенасыщением ее гидроксиапатитом, наиболее выражены при рН слюны 6,5 – 7,5. При подкислении слюны снижается степень насыщения ее гидроксиапатитом. рН 6,0 – 6,2 является критическим, когда слюна из состояния перенасыщенности переходит в ненасыщенное состояние, из минерализующей становится деминералилизирующей жидкостью. Особенно же опасно понижение рН слюны ниже 6,0, так как при этом теряются минерализирующие свойства слюны. С повышением концентрации водородных ионов в ротовой полости повышается проницаемость эмали. Костная ткань принимает участие в нейтрализации нагрузок кислотами и щелочами. Важное практическое значение имеют случаи хронических нагрузок организма кислотами. Так, например, при хронической почечной недостаточности нейтрализация Н+ костным карбонатом вызывает освобождение кальция из кости во внеклеточную жидкость, что может привести к остеопорозу, в том числе и костей челюстно-лицевого аппарата. В настоящее время для выявления нарушений КОС используется метод Аструпа, заключающийся в электрометрическом измерении рН при различных насыщениях крови СО с последующим расчетом остальных показателей по номограмме Сиггаард – Андерсена. Основными из них можно считать следующие. рН крови – величина активной реакции среды – 7,36 – 7,44 (в артериальной крови) и 7,26 – 7,36 (в венозной крови). Напряжение углекислого газа – рСО2 – отражает концентрацию растворенного в плазме артериальной крови углекислого газа – 36 – 46 мм рт. ст. Буферные основания – БО (ВВ) – сумма анионов всех буферных систем крови – 45 – 55 ммоль/л. Стандартный бикарбонат плазмы крови – СБ (SB) – концентрация бикарбоната в плазме крови, приведенная к стандартным условиям – 20 – 27 ммоль/л. Сдвиг буферных оснований – СБО (BE) – отражает метаболический компонент сдвига. Этот показатель изменяется при метаболических сдвигах или при метаболической компенсации газовых сдвигов; в норме равен + 2,0 ммоль/л. Таблица № 1. Характер изменений основных показателей кислотно-основного состояния при ацидозах и алкалозах. Характер изменений основных показателей кислотно-основного состояния при ацидозах и алкалозах 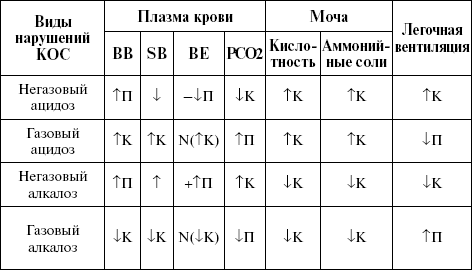 Болезнетворное действие факторов внешней среды Кинетозы. Патогенное действие электрического тока Все многообразие болезнетворных факторов воздействия внешней среды на организм можно разделить на 5 основных групп: механические, физические, химические, биологические и социальные. К механическим факторам относится, в частности, патогенное действие ускорений. Равномерное прямолинейное и вращательное движения не сопровождаются болезнетворными явлениями, но изменение скорости движения (ускорение) может резко изменить состояние организма. Интерес к вопросу о реакциях организма в ответ на воздействие ускорений в последние годы сильно возрос благодаря широкому использованию в народном хозяйстве новых скоростных средств транспорта. Комплекс расстройств, возникающих при передвижении на различных транспортных средствах, можно объединить под общим названием кинетозы, или болезни движения. Кинетозы возникают при качке судна на море (морская болезнь), при полете на самолете (воздушная болезнь), при вращении на карусели, на качелях и т. п. В этих условиях на организм действуют ускорения порядка 1 – 2 g. Большое значение приобрела проблема кинетозов в связи с развитием космических полетов. Симптомокомплекс кинетозов складывается из четырех видов реакций, которые у разных людей проявляются по разному: 1) двигательные реакции, изменение тонуса поперечнополосатой мускулатуры; 2) вегетативные расстройства, проявляющиеся побледнением, холодным потом, отсутствием аппетита, тошнотой, рвотой, брадикардией; 3) сенсорные реакции, характеризующиеся головокружением, нарушением пространственной ориентации; 4) психические расстройства (депрессивные состояния, сонливость, нарушение внимания, галлюцинации и др.). Эти изменения имеют, в основном, рефлекторный характер и обусловлены воздействием на различные рецепторы: 1) вестибулярный анализатор (механорецепторы отолитового аппарата, рецепторы полукружных каналов); 2) проприорецепторы мышц, сухожилий; 3) зрительные рецепторы; 4) рецепторы слизистых и серозных оболочек органов брюшной полости. Лабиринтная гипотеза патогенеза кинетозов является в настоящее время господствующей. Подтверждает эту теорию ряд клинических наблюдений. Так, у глухонемых, например, симптомокомплекс кинетозов не обнаруживается. У детей до 2 лет возбудимость некоторых анализаторов, в том числе и вестибулярного, понижена, поэтому у них также не обнаруживается проявлений кинетозов; напротив, после 40 лет чувствительность к изменению ускорений повышается. Согласно другой точке зрения, кинетозы представляют собой следствие нарушений взаимодействия различных анализаторов. Эта концепция включает внутрилабиринтный конфликт как частный случай. Среди физических факторов, воздействию которых наиболее часто подвергается организм, можно выделить электрический ток. Поражения, возникающие от воздействия электротока, относятся к особому виду травм. В отличие от всех прочих поражений, наносимых организму механическими, химическими и другими физическими агентами, электричество действует на человека не только при соприкосновении, но и косвенно. Более того, электричество может поразить человека на расстоянии – через дуговой контакт и шаговое напряжение. Биологическое действие электрического тока определяется его физическими параметрами, а также состоянием организма. Считается, что патогенный эффект зависит главным образом от силы тока. При силе тока 15 – 20 мА возникают сильные судороги мышц («неотпускающий ток»), при 50 – 100 мА – фибрилляция сердца и паралич дыхания. Ток, превышающий 100 мА, для человека считается смертельным. Патогенное воздействие электрического тока тем сильнее, чем выше его напряжение. Переменный ток ниже 40 В считается безвредным, ток до 100 В – условно патогенным, свыше 200 В – абсолютно патогенным. Наиболее опасен переменный ток с частотой 40 – 60 Гц; с увеличением частоты поражающее действие его уменьшается. Повреждающий эффект зависит также от сопротивления тела и отдельных его тканей электрическому току. Сопротивление колеблется от сотен КОм до нескольких МОм, в зависимости от кровенаполнения органов, степени увлажнения, огрубения и загрязнения кожи, а также от функциональной активности ЦНС. Наибольшим сопротивлением току обладает неповрежденная кожа, наименьшим – кровь и лимфа. Патогенный эффект электрического тока зависит от направления прохождения («петли» тока). Особенно опасно прохождение тока через область сердца и головной мозг. Опасность возрастает с увеличением времени прохождения тока через организм. Повышение обмена веществ, глубокая гипоксия, утомление, кровопотеря снижают резистентность организма к электрическому току; истощение, сердечно-сосудистые заболевания, status thymico-lymphaticus также увеличивают тяжесть поражения электрическим током. Эмоциональное напряжение, ожидание действия тока, гипероксия повышают устойчивость организма к повреждающему действию электротока. Повреждения, возникающие в организме при действии электротока, слагаются из местных изменений (электрические знаки, ожоги, электролиз) и общих проявлений реакции организма на травму (потеря сознания, остановка дыхания, фибрилляция желудочков сердца, изменение кровяного давления, ишемия миокарда, сокращение скелетных мышц и т. д.). При воздействии электрического тока, если не развивается фибрилляция и не останавливается дыхание, может возникнуть электрический шок за счет резкого болевого раздражения рецепторов, нервных стволов, болезненных судорог мышц и спазма сосудов. Электрический ток, проходящий через организм, оказывает электрохимическое, электротермическое и возбуждающее действие. У постоянного тока наиболее выражены электрохимический и тепловой эффекты. Электрохимическое действие проявляется в электролизе, который приводит к поляризации клеточных мембран, изменению функционального состояния клеток, коагуляции белков, газообразованию, накоплению токсических продуктов обмена. Тепловой эффект проявляется ожогами, которые чаще возникают в месте действия. Возбуждающий эффект у постоянного тока наблюдается только в моменты замыкания (на катоде) и размыкания (на аноде). У низкочастотного переменного тока преобладают тепловое действие и возбуждающий эффект. В результате возбуждающего действия происходят сокращение поперечнополосатых и гладких мышц, активация рецепторов, спазм голосовых связок и т. д. Возбуждающий эффект наблюдается в течение всего времени экспозиции тока. Электрохимическое действие у переменного низкочастотного тока выражено незначительно. Переменный высокочастотный ток, в частности УВЧ, используемый в медицине, не обладает возбуждающим и электрохимическим эффектом, но оказывает глубокое прогревающее действие. При длительном же воздействии высокочастотных токов наблюдается патогенный их эффект на организм, проявляющийся в утомлении, тахикардии, аритмии сердца, изменениях со стороны ЦНС и др. Дети нередко подвергаются действию электротока, что связано со слабым контролем со стороны взрослых за детскими играми, недостаточным ограждением бытовых электроприборов. У детей раннего возраста отмечена пониженная резистентность к действию различных факторов внешней среды, в том числе и к электрическому току. Это обусловлено низкой лабильностью нервной системы, что в свою очередь определяет ограниченные возможности приспособления детского организма к колебаниям условий среды. ЛЕКЦИЯ № 4. ТРАВМАТИЧЕСКИЙ ШОК Травматический шок – острый нейрогенный фазный патологический процесс, развивающийся при действии чрезвычайного травмирующего агента и характеризующийся развитием недостаточности периферического кровообращения, гормонального дисбаланса, комплекса функциональных и метаболических расстройств. В патогенезе травматического шока играют роль три основных фактора – нейрогенный, крово– и плазмопотеря и токсемия. В динамике травматического шока различают эректильную и торпидную стадии. В случае неблагоприятного течения шока наступает терминальная стадия. Эректильная стадия шока непродолжительная, длится несколько минут. Внешне проявляется речевым и двигательным беспокойством, эйфорией, бледностью кожных покровов, частым и глубоким дыханием, тахикардией, некоторым повышением артериального давления. В этой стадии происходят генерализованное возбуждение центральной нервной системы, чрезмерная и неадекватная мобилизация всех приспособительных реакций, направленных на устранение возникших нарушений. Пусковым фактором в развитии эректильной фазы шока является мощная болевая и неболевая афферентная импульсация из поврежденных тканей. Афферентная импульсация достигает ретикулярной формации ствола мозга и приводит ее в сильное возбуждение. Отсюда процесс возбуждения иррадиирует в кору, подкорковые центры, продолговатый мозг и спинной мозг, приводя к дезинтеграции деятельности центральной нервной системы, вызывая чрезмерную активацию симпатоадреналовой и гипоталамо-гипофизарно-надпочечниковой систем. Наблюдается массивный выброс адреналина, АКТГ, вазопрессина, глюкокортикоидов и других гормонов. Избыточное освобождение катехоламинов вызывает спазм артериол, в которых преобладают ?-адренорецепторы, в частности, в сосудах кожи, мышц, кишечника, печени, почек, т. е. органов, которые для выживания организма во время действия шокогенного фактора имеют меньшее значение. Одновременно с периферической вазоконстрикцией возникает выраженная централизация кровообращения, обеспечиваемая дилатацией сосудов сердца, мозга, гипофиза. Централизация кровообращения в начальной фазе шока носит адаптационный характер, обеспечивая в достаточном объеме, почти близком к обычному, кровоток в сосудах сердца и головного мозга. Однако если в дальнейшем не происходит быстрой нормализации объема циркулирующей крови, то она приводит к выраженной гипоксии в тех органах, в которых наступает продолжительное ограничение кровотока. Эректильная фаза шока быстро переходит в торпидную. В основе трансформации эректильной стадии в торпидную лежит комплекс механизмов: прогрессирующее расстройство гемодинамики, циркуляторная гипоксия, приводящая к выраженным метаболическим расстройствам, дефицит макроэргов, образование тормозных медиаторов в структурах ЦНС, в частности, ГАМК, простагландинов типа Е, повышенная продукция эндогенных опиоидных нейропептидов. Торпидная фаза травматического шока наиболее типичная и продолжительная, она может длиться от нескольких часов до двух суток. Для нее характерны заторможенность пострадавшего, адинамия, гипорефлексия, диспноэ, олигурия. Во время этой фазы наблюдается торможение активности центральной нервной системы. В развитии торпидной стадии травматического шока в соответствии с состоянием гемодинамики могут быть выделены две фазы – компенсации и декомпенсации. Фаза компенсации характеризуется стабилизацией артериального давления, нормальным или даже несколько сниженным центральным венозным давлением, тахикардией, отсутствием гипоксических изменений в миокарде (по данным ЭКГ), отсутствием признаков гипоксии мозга, бледностью слизистых оболочек, холодной влажной кожей. Для фазы декомпенсации характерны прогрессирующее уменьшение МОК, дальнейшее снижение артериального давления, развитие ДВС-синдрома, рефрактерность микрососудов к эндогенным и экзогенным прессорных аминам, анурия, декомпенсированный метаболический ацидоз. Стадия декомпенсации является прологом терминальной фазы шока, которая характеризуется развитием необратимых изменений в организме, грубыми нарушениями обменных процессов, массивной гибелью клеток. Характерной особенностью травматического шока является развитие патологического депонирования крови. Касаясь механизмов патологического депонирования крови, следует отметить, что они формируются уже в эректильной фазе шока, достигая максимума в торпидной и терминальной стадиях шока. Ведущими факторами патологического депонирования крови являются спазм сосудов, циркуляторная гипоксия, формирование метаболического ацидоза, последующая дегрануляция тучных клеток, активация калликреин-кининовой системы, образование вазодилатирующих биологически активных соединений, расстройство микроциркуляции в органах и тканях, характеризующихся изначально длительным спазмом сосудов. Патологическое депонирование крови приводит к выключению из активной циркуляции значительной части крови, усугубляет несоответствие между объемом циркулирующей крови и емкостью сосудистого русла, становясь важнейшим патогенетическим звеном расстройства кровообращения при шоке. Важную роль в патогенезе травматического шока играет плазмопотеря, которая обусловливается повышением проницаемости сосудов вследствие действия кислых метаболитов и вазоактивных пептидов, а также возрастанием внутрикапиллярного давления из-за застоя крови. Плазмопотеря приводит не только к дальнейшему дефициту объема циркулирующей крови, но и вызывает изменения реологических свойств крови. При этом развиваются явления агрегации клеток крови, гиперкоагуляция с последующим формированием ДВС-синдрома, образуются капиллярные микротромбы, полностью прерывающие ток крови. Кризис микроциркуляции, прогрессирующая недостаточность кровообращения и дыхания приводят к развитию тяжелой гипоксии, которая в дальнейшем определяет тяжесть шокового состояния. В условиях прогрессирующей циркуляторной гипоксии возникают дефицит энергообеспечения клеток, подавление всех энергозависимых процессов, выраженный метаболический ацидоз, повышение проницаемости биологических мембран. Энергии не хватает для обеспечения функций клеток и, прежде всего, таких энергоемких процессов, как работа мембранных насосов. Натрий и вода устремляются в клетку, а калий выделяется из нее. Развитие отека клетки и внутриклеточного ацидоза приводит к повреждению лизосомальных мембран, высвобождению лизосомальных ферментов с их литическим действием на различные внутривнеклеточные структуры. Денатурированные белки и продукты распада нежизнеспособных тканей начинают оказывать токсическое действие. Кроме того, при шоке проявляют токсическое действие многочисленные биологически активные вещества, в избытке поступающие во внутреннюю среду организма (гистамин, серотонин, кинины, свободные радикалы, креатинин, мочевина и др.). Таким образом, по мере прогрессированил шока, вступает в действие еще один ведущий патогенетический фактор – эндотоксемия. Последняя усиливается также за счет поступления токсических продуктов из кишечника, поскольку гипоксия уменьшает барьерную функцию кишечной стенки. Определенное значение в развитии эндотоксемии имеет нарушение антитоксической функции печени. Эндотоксемия наряду с выраженной клеточной гипоксией, обусловленной кризисом микроциркуляции, перестройкой метаболизма тканей на анаэробный путь и нарушением ресинтеза АТФ, играет важную роль в развитии явлений необратимого шока. Течение травматического шока в раннем детском возрасте обладает рядом характерных особенностей, определяемых реактивностью детского организма. Чувствительность к механической травме детей раннего возраста выше, чем взрослых, и поэтому одинаковая по тяжести и локализации травма обусловливает у них развитие более тяжелого травматического шока. Тяжелая механическая травма у детей вызывает более резкие, чем у взрослых, нарушения кислотно-основного состояния. Одной из особенностей травматического шока у детей является развитие ранней и тяжелой гипотермии. У многих детей температура тела снижается до 34 – 35 °С, что объясняется возрастными особенностями функционирования центра терморегуляции. ЛЕКЦИЯ № 5. НАРУШЕНИЕ РЕГИОНАРНОГО КРОВОТОКА К типовым расстройствам регионарного кровообращения относятся: 1) артериальная и венозная гиперемии; 2) ишемия; 3) стаз; 4) тромбоз; 5) эмболия; 6) кровотечение и кровоизлияние. В зависимости от продолжительности развития расстройства кровотока могут быть: 1) преходящими; 2) стойкими; 3) необратимыми. По степени распространенности расстройства кровотока могут носить: 1) диффузный; 2) генерализованный; 3) местный локальный характер. Артериальная гиперемия Артериальной гиперемией (греч. «hyper» – сверх, «haema» – кровь) называется состояние повышенного кровенаполнения органа и ткани, возникающее в результате усиленного притока крови к ним по расширенным артериям. Артериальная гиперемия может быть местной и общей. Общее артериальное полнокровие развивается при значительном увеличении объема циркулирующей крови – например, при эритроцитозе. Патологическая артериальная гиперемия возникает также в условиях гипертермии при перегревании организма, при лихорадке у больных с инфекционными заболеваниями, при быстром падении барометрического давления. Артериальная гиперемия может носить острый, преходящий характер, может быть часто повторяющейся, хронической. Различают физиологические и патологические артериальные гиперемии. При физиологической артериальной гиперемии усиление кровотока адекватно возросшим потребностям органа или ткани в кислороде и энергетических субстратах. Примерами физиологической артериальной гиперемии могут служить рабочая гиперемия, когда к усиленно работающему органу увеличивается приток крови, и гиперемия лица, появляющаяся при чувстве радости, гнева, стыда. Патологическая артериальная гиперемия возникает вне зависимости от метаболических потребностей органа. По особенностям этиологических факторов и механизмов развития выделяют следующие разновидности патологических артериальных гиперемий: 1) нейропаралитическую; 2) нейротоническую; 3) постишемическую; 4) вакатную; 5) воспалительную; 6) коллатеральную; 7) гипермию вследствие артериовенозного свища. В основе патогенеза артериальных гиперемий лежат следующие механизмы: 1) миопаралитический; 2) нейрогенный (ангионевротический). Миопаралитический механизм связан со снижением миогенного тонуса сосудов под влиянием метаболитов (лактата, пуринов, пировиноградной кислоты и др.), медиаторов, внеклеточного увеличения концентрации калия, водорода и других ионов, уменьшения содержания кислорода. Это самый частый механизм развития артериальной гиперемии. Он лежит в основе постишемического, воспалительного, физиологического рабочего артериального полнокровия. Сущность нейрогенного механизма состоит в изменении нейрогенных констрикторных и дилататорных влияний на сосуды, приводящих к снижению нейрогенного компонента сосудистого тонуса. Данный механизм лежит в основе развития нейротонической, нейропаралитической гиперемии, а также воспалительного артериального полнокровия при реализации аксон-рефлекса. Нейротоническая артериальная гиперемия возникает при повышении тонуса парасимпатических или симпатических холинергических сосудорасширяющих нервов или при раздражении их центров опухолью, рубцом и т. д. Этот механизм наблюдается только в некоторых тканях. Под влиянием симпатических и парасимпатических вазодилататоров артериальная гиперемия развивается в поджелудочной и слюнной железах, языке, кавернозных телах, коже, скелетных мышцах и др. Постишемическая артериальная гиперемия представляет собой увеличение кровотока в органе или ткани после временного прекращения кровообращения. Подобная артериальная гиперемия возникает, в частности, после снятия жгута с конечности, быстрого удаления асцитической жидкости. Реперфузия способствует не только положительным изменениям в ткани. Приток большого количества кислорода и усиленное его использование клетками приводят к интенсивному образованию перекисных соединений, активации процессов перекисного окисления липидов и, как следствие, прямому повреждению биологических мембран и свободно-радикальному некробиозу. Вакатная (лат. «vacuus» – пустой) гиперемия наблюдается при уменьшении барометрического давления над какой-либо частью тела. Данный вид гиперемии развивается при быстром освобождении от сдавления сосудов брюшной полости, например, при стремительном разрешении при родах, удалении опухоли, сдавливающей сосуды, или быстрой эвакуации асцитической жидкости. Воспалительная артериальная гиперемия возникает под действием вазоактивных веществ (медиаторов воспаления), вызывающих резкое снижение базального тонуса сосудов, а также вследствие реализации в зоне альтерации нейротоничеекого, нейропаралитического механизмов и аксон-рефлекса. Коллатеральная артериальная гиперемия носит приспособительный характер и развивается вследствие рефлекторного расширения сосудов коллатерального русла при затруднении притока крови по магистральной артерии. Гиперемия вследствие артериовенозного свища может развиться при повреждении артериальных и венозных сосудов в результате образования соустья между артерией и веной. При этом артериальная кровь под давлением устремляется в венозное русло, обеспечивая артериальное полнокровие. Для артериальной гиперемии характерны следующие изменения микроциркуляции: 1) расширение артериальных сосудов; 2) увеличение линейной и объемной скоростей кровотока в микрососудах; 3) повышение внутрисосудистого гидростатического давления, увеличение количества функционирующих капилляров; 4) усиление лимфообразования и ускорение лимфообращения; 5) уменьшение артериовенозной разницы по кислороду. К внешним признакам артериальной гиперемии относится покраснение зоны гиперемии, обусловленное расширением кровеносных сосудов, увеличением количества функционирующих капилляров и повышением содержания оксигемоглобина в венозной крови. Артериальная гиперемия сопровождается местным повышением температуры, что объясняется усиленным притоком более теплой артериальной крови и повышением интенсивности обменных процессов. Вследствие возрастания крове– и лимфонаполнения зоны гиперемии происходит увеличение тургора (напряжения) и объема гиперемированной ткани. Венозная гиперемия Венозная гиперемия – это состояние повышенного кровенаполнения органа или ткани, обусловленное затрудненным оттоком крови по венам. Венозное полнокровие может быть местным и распространенным. Местное венозное полнокровие возникает при затруднении оттока крови по крупным венозным стволам вследствие закупорки их тромбом, эмболом или при сдавлении вены извне опухолью, рубцом, отеком и т. д. Условием, способствующим венозному застою, является длительное нефизиологическое положение той или иной части тела, неблагоприятное для местного оттока крови. При этом формируется гипостаз – гравитационная венозная гиперемия. Причинами распространенного венозного полнокровия наиболее часто являются: 1) недостаточность функции сердца при ревматических и врожденных пороках его клапанов, миокардитах, инфаркте миокарда; 2) декомпенсация гипертрофированного сердца; 3) уменьшение присасывающего действия грудной клетки при экссудативном плеврите, гемотораксе и т. д. По темпу развития и длительности существования данная патология может носить острый и хронический характер. Длительная венозная гиперемия возможна только при недостаточности коллатерального венозного кровообращения. Микроциркуляторные расстройства при венозной гиперемии характеризуются: 1) расширением капилляров и венул; 2) замедлением кровотока по сосудам микроциркуляторного русла вплоть до стаза; 3) утратой деления кровотока на осевой и плазматический; 4) повышением внутрисосудистого давления; 5) маятникообразным или толчкообразным движением крови в венулах; 6) уменьшением интенсивности кровотока в области гиперемии; 7) нарушением лимфообращения; 8) увеличением артериовенозной разницы по кислороду. К внешним признакам венозной гиперемии относятся: 1) увеличение, уплотнение органа или ткани; 2) развитие отека; 3) возникновение синюшности, т. е. цианотичной окраски. При остром венозном полнокровии может наблюдаться выход эритроцитов из мелких сосудов в окружающие ткани. При скоплении значительного их количества в слизистых и серозных оболочках, коже формируются мелкие, точечные кровоизлияния. Вследствие усиления транссудации в тканях накапливается отечная жидкость. Количество ее может быть весьма значительным в подкожной клетчатке (анасарка), плевральных полостях (гидроторакс), брюшной полости (асцит), перикарде (гидроперикард), желудочках мозга (гидроцефалия). В условиях гипоксии в клетках паренхиматозных органов развиваются зернистая и жировая дистрофия, мукоидное набухание межуточного вещества. Эти изменения носят, как правило, обратимый характер и при устранении причины острое венозное полнокровие завершается полным восстановлением структуры и функции тканей. При хроническом венозном полнокровии происходят развитие дистрофических процессов в тканях, атрофия паренхиматозных элементов с одновременным заместительным разрастанием клеток стромы и накоплением в ней коллагеновых волокон. Необратимое склерозирование и уплотнение органа сопровождается нарушением его функций и называется цианотической индурацией. Отек Отек – типовой патологический процесс, заключающийся в избыточном накоплении внеклеточной тканевой жидкости в интерстициальном пространстве. По этиологии, патогенезу, распространенности отеки подразделяются на: 1) системные (общие); 2) местные (локальные). Системные отеки возникают вследствие нарушения ведущих механизмов регуляции водно-солевого обмена, что возможно при заболеваниях сердца, почек, печени, желудочно-кишечного тракта и др. В зависимости от локализации различают следующие виды отеков: 1) анасарка – в подкожной жировой клетчатке; 2) водянка – в серозных полостях; 3) гидроперикард – накопление отечной жидкости в сердечной сорочке; 4) гидроторакс – в плевральной полости; 5) асцит – в брюшной полости; 6) гидроцеле – в полости влагалищной оболочки яичка; 7) гидроцефалия – в желудочках мозга. В соответствии с особенностями этиологического фактора и механизмов развития отеки могут быть: 1) воспалительного характера, обусловленные экссудацией; 2) невоспалительного характера, связанные с усилением процесса транссудации и/или нарушением лимфатического дренажа. В зависимости от ведущего фактора, определяющего развитие отека, выделяют: 1) застойные (механические) отеки, обусловленные нарушением крово– и лимфооттока и повышением гидростатического давления в микрососудах; 2) онкотические – вследствие уменьшения величины коллоидно-осмотического давления плазмы крови; 3) мембраногенные – при повышении проницаемости капиллярной стенки; 4) отеки, связанные с активной задержкой в тканях электролитов, преимущественно натрия, и воды; 5) лимфогенные, возникающие при застое лимфы. В зависимости от ведущей причины развития местные отеки можно подразделить на: 1) воспалительные; 2) гемодинамические; 3) лимфодинамические. В основе патогенеза любого местного отека лежит нарушение равновесия Старлинга, которое сводится к возрастанию внутрисосудистого гидростатического давления, снижению онкотического градиента, повышению проницаемости сосудистых стенок, либо комбинации этих механизмов. Так, воспалительный отек связан с развитием в очаге воспаления экссудации, т. е. выхода жидкой части крови из сосудистого русла в очаг воспаления. Экссудация обусловлена: 1) повышением проницаемости сосудистой стенки под влиянием избыточных концентраций вазоактивных соединений, лизосомальных ферментов, ионов водорода, накапливающихся в зоне альтерации; 2) возрастанием гидростатического давления в сосудах микроциркуляторного русла и увеличением площади фильтрации жидкой части крови в условиях венозного застоя, снижением внутрисосудистого онкотического давления при одновременном повышении онкотического давления в тканях; 3) увеличением коллоидно-осмотического давления в тканях и увеличением гидрофильности тканей; 4) активацией процесса цитопемсиса эндотелием сосудов, т. е. захватом эндотелиальными клетками мельчайших капелек плазмы и переносом их за пределы сосуда в воспаленную ткань. Лимфодинамический отек возникает при первичном нарушении лимфооттока, что наблюдается при врожденном дефекте развития лимфатических сосудов, удалении регионарных лимфоузлов, при закупорке, формировании лимфоэктазий и воспалительном поражении лимфатических узлов. Развитию отеков общего характера способствуют следующие факторы. 1. Гиперфункция ренин-ангиотензин-альдостероновой системы и общий избыток натрия в организме (сердечная недостаточность, воспалительные или ишемические поражения почек). 2. Недостаточность образования предсердного натрийуретического фактора (ПНУФ). Как известно, ПНУФ представляет собой комплекс атриопептидов I, II, III, которые синтезируются клетками правого предсердия и его ушка. ПНУФ обладает эффектами, противоположными эффектам альдостерона и антидиуретического гормона, увеличивая выделение с мочой воды и натрия. 3. Снижение онкотического давления плазмы крови вследствие утраты онкотически активных белков (потеря белков при нефротическом синдроме, ожоговой плазморрее, при длительной рвоте и т. д.). 4. Повышение гидростатического давления в обменных сосудах микроциркуляторного русла (застойные явления при сердечной недостаточности, расстройствах водно-электролитного баланса различной этиологии и т. д.). 5. Повышение проницаемости сосудистых стенок (системное действие биологически активных веществ, токсических и ферментных факторов патогенности микроорганизмов, токсинов неинфекционного происхождения и т. д.). 6. Повышение гидрофильности тканей (при нарушениях электролитного баланса, при отложении мукополисахаридов в коже и подкожной клетчатке при микседеме, при нарушениях перфузии тканей кровью в условиях венозного застоя и т. д.). Накопление отечной жидкости в рыхлой подкожной соединительной ткани происходит, прежде всего, под глазами, на тыльной поверхности кистей рук, стоп, на лодыжках, а затем постепенно распространяется на все туловище. Кожа становится бледной, натянутой, морщины и складки разглаживаются. Отечная жировая клетчатка становится бледно-желтой, блестящей, слизеподобной. Клинически начальному отеку с отрицательным тканевым давлением жидкости соответствуег симптом образования ямки при нажатии на отечную ткань. Если ямка от нажатия не образуется – давление в ткани положительное, что соответствует далеко зашедшему, «напряженному» отеку. Тромбоз Тромбоз – прижизненное местное пристеночное образование в сосудах или сердце плотного конгломерата из форменных элементов крови и стабилизированного фибрина, т. е. тромба. Тромбоз – физиологический защитный процесс, направленный на предотвращение кровотечения при травме тканей, на укрепление стенок аневризм, на ускорение стягивания и заживления ран. Однако, если тромбоз избыточен, недостаточен, либо утратил свой обязательно местный ограниченный характер, возможно развитие тяжелой патологии. Тромбы подразделяются на белые, красные и смешанные. Тромбоз как естественный способ остановки кровотечения отражает характер взаимодействия механизмов системы гемостаза и фибринолиза. Принято выделять три основных звена гемостаза: 1) сосудистое звено – гемостатические механизмы сосудистой стенки, направленные на спазм поврежденного сосуда и запуск тромбообразования и свертывания крови; 2) клеточное (тромбоцитарно-лейкоцитарное) звено, которое обеспечивает формирование белого тромба; 3) фибриновое звено – система свертывания, обеспечивающая образование фибрина, в результате чего формируются красные и смешанные тромбы. Все три звена гемостаза включаются одновременно и обеспечивают остановку кровотечения и восстановление целостности сосудистой стенки. Белый тромб формируется за 2 – 5 мин. Образование богатого фибрином красного тромба требует 4 – 9 мин. Процесс тромбообразования начинается с постепенного формирования белого тромба. Белые тромбы обеспечивают остановку капиллярного кровотечения. Красный тромб формируется в условиях преобладания коагуляции над агглютинацией, при быстром свертывании крови и медленном кровотоке. Он способен остановить кровотечение из артериальных и венозных сосудов. Красный тромб состоит из головки, представляющей собой аналог белого тромба, слоистого тела, в котором чередуются тромбоцитарные и фибриновые отложения, и фибринового хвоста, улавливающего эритроциты. Смешанными тромбами называют слоистые тромбы с несколькими агглютинационными белыми головками. Кроме того, выделяют особые виды тромбов, образующихся при определенных условиях: 1) септический тромб, образующийся при инфекционных воспалительных поражениях сосудов (флебиты, васкулиты); 2) опухолевый тромб, формирующийся из агглютинированных тромбоцитов и лейкоцитов на клетках проросшей в сосуд опухоли; 3) шаровидный тромб, имеющий смешанный характер и образующийся на основе оторвавшегося пристеночного тромба при нарушениях внутрисердечной гемодинамики вследствие митрального стеноза; 4) вегетации – тромбы, наслаивающиеся на пораженные эндокардитом клапаны сердца; 5) марантический тромб – красный тромб, который формируется при венозном застое на фоне дегидратации и сгущения крови. Тромб следует отличать от кровяного сгустка. Истинный тромб всегда формируется только прижизненно внутри сосудов и прочно спаян с сосудистой стенкой. Сгустки же могут образовываться не только прижизненно, но и в условиях in vitro (посмертно), не только в просвете сосуда, но также и в полостях и в тканях на месте гематом. Сгустки крови в сосудах лежат свободно или сцеплены с сосудистой стенкой рыхло, не имеют структурированности, свойственной тромбам. В основе активации тромбообразования при различных патологических процессах лежит триада Вирхова: повреждение эндотелия сосудистой стенки, замедление кровотока, а также активация коагуляционного гемостаза. Указанный каскад реакций может индуцироваться эндотоксинами грамотрицательных бактерий, экзотоксинами, гипоксией, избыточным накоплением водородных ионов, биогенными аминами, кининами, лейкотриенами, проэтагландинами, свободными радикалами и многими цитокинами, продуцируемыми в избытке нейтрофилами, моноцитами, лимфоцитами. Последствия тромбоза могут быть разнообразны. С одной стороны, тромбоз является защитным механизмом, направленным на остановку кровотечения при повреждении или разрыве сосуда. С другой стороны, тромбоз, возникающий при различных патологических состояниях, ведет к развитию нарушений местного кровобращения, нередко с тяжелыми последствиями для организма. Характер циркуляторных расстройств и степень нарушения функции opганов при тромбозах могут быть различными и зависят от локализации тромба, скорости его образования, возможностей коллатерального кровообращения в данном месте. Эмболия Эмболией называется закупорка кровеносного или лимфатического сосуда частицами, приносимыми с током крови или лимфы, и обычно не встречающимися в крово– и лимфотоке. По направлению движения эмбола различают: 1) ортоградную; 2) ретроградную; 3) парадоксальную эмболии. Ортоградная эмболия встречается чаще всего и характеризуется продвижением эмбола по направлению тока крови. При ретроградной эмболии эмбол движется против тока крови под действием собственной силы тяжести. Это происходит в венозных сосудах, по которым кровь течет снизу вверх. Парадоксальная эмболия имеет ортоградное направление, но возникает вследствие дефектов межпредсердной или межжелудочковой перегородки, когда эмбол имеет возможность миновать разветвления легочной артерии и оказаться в большом круге кровообращения. Эмболия может быть одиночной и множественной. В зависимости от локализации различают: 1) эмболии лимфатических и кровеносных сосудов; 2) эмболии малого круга кровообращения; 3) эмболии большого круга кровообращения; 4) эмболии системы воротной вены. При эмболии большого круга кровообращения источником эмболов являются патологические процессы (тромбоэндокардиты, инфаркт миокарда, изъязвления атеросклеротических бляшек) в легочных венах, левых полостях сердца, аорте, артериях большого круга кровообращения. Эмболии большого круга кровообращения сопровождаются серьезными расстройствами кровообращения вплоть до развития очагов некроза в органе, сосуд которого закупорен тромбом. Эмболия малого круга кровообращения является результатом заноса эмболов из правой половины сердца и вен большого круга кровообращения. Для эмболии малого круга кровообращения характерны внезапность возникновения, быстрота нарастания чрезвычайно тяжелых клинических проявлений. По характеру эмбола различают экзогенные и эндогенные эмболии. К экзогенным эмболиям относятся: 1) воздушная; 2) газовая; 3) микробная; 4) паразитарная. К эндогенным эмболиям относятся: 1) тромбоэмболии; 2) жировая; 3) тканевая. Воздушная эмболия возникает вследствие попадания в сосудистую систему воздуха из окружающей среды. Причинами воздушной эмболии могут быть повреждения крупных вен шеи, грудной клетки, синусов твердой мозговой оболочки, нейрохирургические операции со вскрытием венозных синусов, искусственное кровообращение, лечебные и диагностические пункции легких, газоконтрастные рентгенологические исследования, лапароскопические операции и т. д. Газовая эмболия связана с выделением в крови пузырьков растворенных в ней газов (азота и гелия) при быстром переходе от высокого атмосферного давления к обычному или от нормального к пониженному. Такая ситуация может возникнуть при внезапной декомпрессии, например, при быстром подъеме водолаза со значительной глубины (кессонная болезнь), при разгерметизации барокамеры или кабины космического корабля и т. п. Микробная эмболия имеет место при септикопиемиях, когда в кровотоке находится большое количество микроорганизмов. Микробная эмболия может быть причиной развития метастатических абсцессов. Паразитарная эмболия встречается при гельминтозах. Так, например, при аскаридозе возможна эмболия сосудов легких. Жировая эмболия наступает при закупорке сосудов эндогенными липопротеидными частицами, продуктами агрегации хиломикронов или экзогенными жировыми эмульсиями и липосомами. При истинной жировой эмболии имеет место высокий уровень свободных жирных кислот в крови, которые обладают аритмогенным действием. Тканевая эмболия подразделяется на: 1) амниотическую; 2) опухолевую; 3) адипоцитарную. Эмболия околоплодными водами приводит к закупорке легочных сосудов конгломератами клеток, взвешенных в амниотической жидкости, и тромбоэмболами, образующимися под действием содержащихся в ней прокоагулянтов. Опухолевая эмболия представляет собой сложный процесс гематогенного и лимфогенного метастазирования злокачественных новообразований. Опухолевые клетки образуют в кровотоке конгломераты с тромбоцитами за счет продукции муцинов и адгезивных поверхностных белков. Тканевая и, в частности, адипоцитарная эмболия может быть результатом травм, когда частички размозженных тканей попадают в просвет поврежденных сосудов. Эмболия инородными телами встречается довольно редко и возникает при ранениях или медицинских инвазивных процедурах. Разновидность эндогенной эмболии – тромбоэмболия – возникает вследствие закупорки сосудов оторвавшимися тромбами или их частицами. Тромбоэмболия является следствием тромбоза или тромбофлебита различных отделов венозной системы организма. Одной из наиболее тяжелых форм тромбоэмболии является тромбэмболия легочной артерии (ТЭЛА), частота встречаемости которой в клинической практике постоянно увеличивается в последние годы. Причиной ТЭЛА в 83 % случаев является флеботромбоз центральных и периферических сосудов, в частности, подвздошной, бедренной, подключичной вен, глубоких вен голени, вен таза и др. Характер клинических проявлений и тяжесть последствий ТЭЛА могут зависеть от калибра окклюзированного сосуда, скорости развития процесса и резервов системы фибринолиза. По характеру течения ТЭЛА различают формы: 1) молниеносную; 2) острую; 3) подострую; 4) рецидивирующую. Молниеносная форма характеризуется развитием основных симптомов в течение нескольких минут, острая – в течение нескольких часов, подострая – в течение нескольких дней. По степени поражения легочного сосудистого русла выделяют формы: 1) массивную; 2) субмассивную; 3) форму с поражением мелких ветвей легочной артерии. Массивная форма возникает при эмболии ствола и главных ветвей легочной артерии, т. е. при поражении более 50 % легочного сосудистого русла. При субмассивной эмболии происходит перекрытие долевых ветвей легочной артерии, т. е. менее 50 % сосудистого русла легких. Ишемия Ишемией (греч. «isho» – задерживаю) называется малокровие тканей, вызванное недостаточным или полным прекращением притока артериальной крови. По причинам возникновения и механизмам развития различают несколько видов ишемии: 1) ангиоспастическую, возникающую в результате спазма артерий, обусловленного либо повышением тонуса вазоконстрикторов, либо воздействием на стенки сосудов сосудосуживающих веществ; 2) компрессионную, вызывающуюся сдавлением артерий рубцом, опухолью, наложенным жгутом, излившейся кровью и т. д.; 3) обтурационную, развивающуюся при частичном или полном закрытии просвета артерии тромбом, эмболом, атеросклеротической бляшкой и т. д.; 4) перераспределительную, имеющую место при межрегиональном, межорганном перераспределении крови; 5) обструктивную, возникающую в результате механического разрушения сосудов при травме; 6) ишемию, обусловленную значительным увеличением вязкости крови в мелких сосудах в сочетании с вазоконстрикцией. Перечисленные виды ишемии чаще всего развиваются достаточно быстро и относятся к категории острых. Хроническая ишемия развивается медленно, при постепенном сужении просвета артерий вследствие утолщения их стенок при атеросклерозе, гипертонической болезни, ревматизме. Ишемизированный участок отличается бледностью, уменьшением объема и тургора вследствие нарушения кровенаполнения. Происходит снижение температуры участка ишемии из-за нарушения притока теплой артериальной крови и уменьшения интенсивности обменных процессов. Снижается величина пульсации артерий в результате уменьшения их систолического наполнения. Вследствие раздражения тканевых рецепторов недоокисленными продуктами обмена веществ возникают боли, парестезии. Ишемия характеризуется следующими нарушениями микроциркуляторного кровотока: 1) сужением артериальных сосудов; 2) замедлением тока крови по микрососудам; 3) уменьшением количества функционирующих капилляров; 4) понижением внутрисосудистого гидростатического давления; 5) уменьшением образования тканевой жидкости; 6) понижением напряжения кислорода в ишемизированной ткани. Вследствие нарушения доставки кислорода и субстратов обмена веществ в ишемизированной ткани развиваются обменные, структурные и функциональные нарушения, выраженность которых зависит от следующих факторов: 1) от скорости развития и продолжительности ишемии; 2) от чувствительности тканей к гипоксии; 3) степени развития коллатерального кровотока; 4) предшествующего функционального состояния органа или ткани. Ишемические участки испытывают состояние кислородного голодания, снижается интенсивность обменных процессов, развивается дистрофия паренхиматозных клеток вплоть до их гибели, исчезает гликоген. При продолжительной запредельной ишемии может наступить омертвение ткани. Так, клетки коры головного мозга погибают через 5 – 6 мин после прекращения притока артериальной крови, сердечная мышца выдерживает гипоксию продолжительностью 20 – 25 мин. Инфаркт Инфаркт (лат. infarctus – начиненный, набитый) – это очаг некроза, возникающий в результате прекращения притока крови к органам с функционально конечными сосудами, т. е. или не имеющими, или имеющими крайне недостаточное количество анастомозов. К таким органам относятся головной мозг, легкие, селезенка, почки, печень, тонкий кишечник, где сосуды имеют анастомозы лишь в области микроциркуляторного русла и поэтому при задержке кровотока по основному стволу коллатерали оказываются недостаточными для купирования ишемии в бассейне поврежденного сосуда. Непосредственной причиной инфаркта обычно является тромбоз соответствующей артерии, реже – эмболия; допускается также длительный спазм артерий. Различают следующие разновидности инфарктов в зависимости от различных признаков и механизмов развития: 1) белые и красные; 2) асептические и инфицированные; 3) коагуляционные и колликвационные; 4) пирамидальноконической и неправильной формы. Белые (ишемические) инфаркты возникают в органах с абсолютно или относительно недостаточными коллатералями (почки, сердце, головной и спинной мозг, селезенка, печень) и характеризуются отсутствием вторичного заполнения кровеносных сосудов некротического участка кровью. Красные (геморрагические) инфаркты возникают при вторичном затекании крови в сосуды зоны некроза из коллатералей или через портальные системы и выраженном диапедезе крови (легкие, кишечник, гонады, сетчатка и т. д.). Инфаркты внутренних органов чаще бывают асептическими. Инфицированный инфаркт развивается в случае первичного бактериального обсеменения участка, подвергшегося некрозу (кишечник, легкие) или в результате закупорки сосудов инфицированными тромбами или эмболами. Во всех органах инфаркт развивается по типу коагуляционного некроза с исходом в соединительнотканный рубец. Лишь инфаркты мозга протекают по типу коллимационного некроза, с незначительным участием нейтрофильных лейкоцитов, активацией элементов микроглии и исходом в виде кисты. Форма зоны некроза зависит от характера ветвления кровеносных сосудов. Так, в органах с дихотомическим разветвлением сосудов (селезенка, легкие, почки) зона некроза имеет пирамидально-коническую форму; в сердце, мозге возникает некроз неправильной формы. Стаз Стаз (греч. «stasis» – остановка) – это обратимая остановка кровотока в сосудах микроциркуляторного русла. Стаз может быть вызван уменьшением разницы давлений на протяжении микрососуда или увеличением сопротивления в его просвете. В зависимости от причин и механизмов возникновения различают несколько видов стаза: 1) истинный (капиллярный); 2) ишемический; 3) венозный. Истинный стаз связан со значительным первичным увеличением сопротивления кровотоку в сосудах, которое возникает вследствие нарушения реологических свойств крови (гематокрита, вязкости, деформируемости и клейкости форменных элементов) и внутрикапиллярной агрегации клеток крови. В основе ишемического и венозного стаза лежат дисциркуляторные расстройства: резкое замедление или полное прекращение притока артериальной крови или нарушение оттока венозной крови. При стазе кровоток полностью прекращается, эритроциты склеиваются и образуют агрегаты в виде так называемых «монетных столбиков», вплоть до гомогенизации форменных элементов крови, что получило название «сладж-феномен» (англ. sludge – тина, болото). Исход стаза зависит от его длительности и места возникновения. Кратковременный стаз обратим, при быстром устранении причин стаза движение крови восстанавливается. Длительный стаз приводит к распаду тромбоцитов с последующим выпадением фибрина и образованием тромба, что сопровождается развитием прогрессирующей циркуляторной гипоксии и некроза тканей. Кровотечение Кровотечение, геморрагия (греч. haema – кровь, rhagos – разорвать) – это выход крови из сердца или сосудов. Оно называется наружным, если кровь вытекает во внешнюю среду, и внутренним, когда кровь скапливается в тканях или естественных полостях тела: в плевральных – гемоторакс, в перикарде – гемоперикард, брюшной полости – гемоперитонеум, суставах – гемартроз. По характеру кровоточащего сосуда кровотечения делятся на: 1) артериальные; 2) венозные; 3) капиллярные; 4) смешанные. По механизму нарушения целостности сосудистой стенки выделяют следующие виды кровотечений: 1) per rhexin (лат. «гехо» – разрываю) – кровотечение в результате разрыва стенок сосудов или сердца, которое возникает при механических травмах, некрозе стенок сосудов или сердца, разрыве стенки врожденной или приобретенной аневризмы, при первичных патологических процессах в стенке сосуда (сифилис, атеросклероз и т. д.); 2) per diabrosin (греч. «diabrosin» – разъедание) – кровотечение вследствие разъедания стенок сосудов, т. е. энзиматического переваривания компонентов стенки сосуда при геморрагическом панкреатите, язвенном дефекте желудка или двенадцатиперстной кишки, гнойном расплавлении ткани и т. д.; 3) per diapedesin (греч. «dia» – через, «pedeo» – скачу) – выход эритроцитов через стенки сосудов, неимеющих видимых повреждений; он возникает в области микроциркуляторного русла вследствие повышения проницаемости артериол, венул и капилляров при инфекционных, сосудистых заболеваниях, при поражениях кроветворного аппарата. По внешнему виду различают несколько видов кровоизлияний: 1) петехиальные (мелкие, точечные) возникают путем диапедеза из сосудов мелкого калибра; они часто возникают в коже, слизистых и серозных оболочках при инфекциях, болезнях крови, гипоксии и т. д.; более крупные кровоизлияния называются экхимозы; множественные петехии и экхимозы характеризуются как пурпура; 2) кровоподтеки («синяки») – пластинчатые кровоизлияния в рыхлой подкожной клетчатке, которые возникают при травмах вследствие разрыва мелких сосудов и диапедеза; 3) геморрагическая инфильтрация (суффузия) – обширная по протяженности, характеризуется накоплением крови в межтканевых щелях, «пропитыванием» ткани кровью; 4) гематома – отличается местным разрушением ткани и образованием полости, содержащей кровь и/или сгустки; типично образование гематом в головном мозге при атеросклерозе, ревматизме, гипертонической болезни; механизм образования гематомы может быть сложен. Диссеминированное внутрисосудистое свертывание крови (ДВС-синдром) ДВС-синдром – неспецифический патологический процесс, характеризующийся распространенным свертыванием крови и агрегацией клеток крови в микроциркуляции, ведущим к блокаде микроциркуляции, гипоксии, ацидозу, дистрофии органов, развитию полиорганной недостаточности. ДВС-синдром осложняет самые разнообразные формы патологии: инфаркт миокарда, кардиогенный шок, различные виды злокачественных новообразований, обширные оперативные вмешательства, тяжелую гипоксию, акушерскую патологию (преждевременная отслойка плаценты, эмболия околоплодными водами, внутриутробная гибель плода), переливание несовместимой крови, системную красную волчанку, иммунокомплексные заболевания, цирроз печени. Развитие ДВС-синдрома при септической инфекции является в значительной мере цитокинопосредованным процессом. Диссеминированное внутрисосудистое свертывание крови – динамический патологический процесс, характеризующийся последовательной сменой генерализованной гиперкоагуляции с внутрисосудистым свертыванием крови, агрегацией тромбоцитов, блокадой микроциркуляции и гипокоагуляции с гипофибриногенемией и тромбоцитопенией потребления. Касаясь патогенеза диссеминированного внутрисосудистого свертывания крови, следует отметить общие закономерности его развития, включающего следующие инициирующие механизмы. 1. Первичное поражение сосудистой стенки, десквамация эндотелия, обнажение субэндотелиальных белков (коллагена, тромбоспондина, фибронектина, фактора Виллебранда), обладающих способностью активировать процессы адгезии и агрегации тромбоцитов, активировать XII-фактор Хагемана с последующей активацией внутреннего механизма протромбиназной активности, системы комплемента, фибринолиза, калликреин-кининовой системы. Таким образом, одновременно запускается тромбоцитарное звено системы гемостаза, коагуляционный гемостаз, сочетающийся с дальнейшей деструкцией сосудистой стенки под влиянием вазоактивиых компонентов комплемента, калликреин-кининовой системы. 2. Первичное преимущественное воздействие патогенного фактора на тромбоциты, индукция процессов адгезии, агрегации тромбоцитов, высвобождение из тромбоцитов биогенных аминов, тромбоцитарных факторов свертывания крови, в частности – III и IV, инициирующих образование тромбина с возможной последующей активацией под влиянием тромбина ряда факторов формирования протромбиназы, развития каскада реакций вторичной активации коагуляционного гемостаза. 3. Сочетанное одномоментное воздействие бактериальных, токсических, иммуноаллергических факторов на тромбоцитарно-сосудистое и коагуляционное звенья системы гемостаза. 4. Развитие альтернативных механизмов гемокоагуляции за счет активации моноцитарно-макрофагального и эритроцитарного звеньев системы гемостаза. Как известно, клеточные элементы мононуклеарной фагоцитирующей системы играют исключительно важную роль в эндоцитозе, переработке антигенов и представлению их Т-хелперам в комплексе с 1а-антигеном. Однако следует отметить, что антиген-стимулированные мононуклеарные фагоциты могут синтезировать около 100 различных биологически активных соединений – монокинов, причем среди монокинов имеется группа высокоактивных соединений, регулирующих процессы гемостаза и фибринолиза в случае развития неспецифических патологических реакций, процессов, синдрома системного воздействия воспаления на организм. В физиологических условиях эта группа клеток практически не продуцирует факторов гемокоагуляции и фибринолиза. Таким образом, различные по природе патогенные факторы, вызывающие развитие локального воспалительного процесса и синдром системного воспалительного ответа, вызывают активацию прокоагулянтной системы крови за счет массивного генерализованного повреждения сосудистой стенки, повышения ее адгезивных свойств, активации тромбоцитарного звена системы гемостаза, а в ряде случаев – моноцитарно-макрофагального и эритроцитарного альтернативных путей гемокоагуляции. Естественно, что в каждом конкретном случае патологии можно выявить определенную специфику инициирующих механизмов активации коагуляционного, тромбоцитарно-сосудистого звеньев системы гемостаза. Однако очень быстро в динамике патологии в связи с каскадом взаимомодулирующих реакций гемостаза теряются специфические особенности расстройств коагуляционного потенциала крови; возникает фаза гиперкоагуляции. В развитии ДВС-синдрома следует выделять следующие фазы: I – гиперкоагуляция и агрегация клеток крови; II – переход гиперкоагуляции в гипокоагуляцию; III – стадия глубокой гипокоагуляции, вплоть до полной несвертываемости крови, которая обусловлена потреблением, расщеплением и блокадой ряда факторов свертывания крови, накоплением и циркуляцией продуктов их распада, обладающих антикоагулянтной активностью, а также тромбоцитопенией потребления; IV – восстановительная стадия при благоприятном течении заболевания или формирование полиорганной недостаточности. ДВС-синдром может носить острый, подострый, хронический и рецидивирующий характер. Острая форма возникает при септических инфекциях, обширных оперативных вмешательствах, кровопотере, ожогах, переливании, несовместимой крови и т. д. В случае острого течения фаза гиперкоагуляции чрезвычайно кратковременна. Подострое течение ДВС-синдрома имеет место при почечной недостаточности, злокачественных новообразованиях, лейкозах. Рецидивирующие и хронические формы могут иметь место при раке, системных воспалительных, аутоиммунных заболеваниях. ЛЕКЦИЯ № 6. ВОСПАЛЕНИЕ Сосудистые реакции и эмиграция лейкоцитов в очаге острого воспаления Воспаление – типовой патологический процесс, возникающий в ответ на действие разнообразных альтерирующих факторов и проявляющийся развитием комплекса сосудисто-тканевых изменений. Основными признаками воспаления являются: боль, отек, краснота, повышение температуры и нарушение функции. В зависимости от реактивности организма, воспаление может быть нормергическим, гиперергическим и гипоэргическим. Изменения со стороны тканей протекают в виде альтерации, экссудации и пролиферации. Различают первичную и вторичную альтерацию. Первичная альтерация развивается в зоне действия фактора, характеризующегося повреждением клеточных элементов ткани с последующим освобождением и образованием медиаторов воспаления – гистамина, серотонина, ацетилхолина, лизосомальных гидролитических ферментов, высокоактивных продуктов протеолиза (кининов) и липолиза (простагландинов и лейкотриенов), а также активацией комплемента. Диффузия вышеуказанных субстанций за пределы зоны первичной альтерации сопровождается развитием вторичной альтерации. Действие БАВ, ионов водорода и гидролаз приводит к нарушениям обмена веществ, расстройствам кровообращения и лимфообращения в очаге воспаления. Острое воспаление характеризуется определенной последовательностью сосудистых изменений, проявляющихся развитием спазма сосудов, артериальной и венозной гиперемии, стазом. Спазм сосудов – реакция кратковременная и связана с непосредственным раздражением, альтерирующим фактором вазоконстрикторов и гладких мышц сосудов. Артериальная гиперемия характеризуется умеренным расширением артериол, капилляров, увеличением скорости кровотока, феноменом новообразования капилляров, увеличением объемной скорости кровотока, повышением внутрикапиллярного давления и некоторым усилением фильтрации жидкой части крови. В основе развития воспалительной артериальной гиперемии лежит несколько механизмов: 1) рефлекторный – активация аксон-рефлекса; 2) нейропаралитический – в силу пареза вазоконстрикторов в кислой среде происходит инактивация действия катехоламинов; 3) миопаралитический – за счет снижения базального тонуса сосудов; падение базального тонуса обусловлено действием накапливающихся вазоактивных медиаторов воспаления и ионов водорода, которые расслабляют мышечные элементы стенки артериол и прекапилляров; 4) разрушение соединительной ткани вокруг сосудов – жесткость капилляров в значительной мере (до 93 %) определяется окружающей соединительной тканью; распад соединительной ткани под влиянием лизосомальных гидролаз приводит к снижению механического противодействия растягивающему усилию давления крови внутри сосуда; активная артериальная гиперемия может продолжаться в течение нескольких часов и суток. По мере нарастания воспалительного процесса артериальная гиперемия сменяется венозной. Венозная гиперемия характеризуется дальнейшим расширением сосудов, замедлением кровотока, феноменом краевого стояния лейкоцитов и их эмиграцией, развитием экссудации, нарушением реологических свойств крови. Факторы, влияющие на переход артериальной гиперемии в венозную можно разделить на две группы – внутрисосудистые и внесосудистые. К внутрисосудистым факторам относятся сгущение крови вследствие перехода жидкой части плазмы из крови в воспаленную ткань, набухание эндотелия в кислой среде, пристеночное стояние лейкоцитов, образование микротромбов вследствие агрегации тромбоцитов и увеличения свертываемости крови. Внесосудистые факторы – это избыточное накопление в очаге воспаления медиаторов воспаления с сосудорасширяющим действием, ионов водорода, сдавление экссудатом стенок вен и лимфатических сосудов. Развивающийся в процессе венозной гиперемии престаз сменяется стазом. Важнейшим признаком венозной гиперемии является эмиграция лейкоцитов, т. е. выход форменных элементов белой крови за пределы сосудистого русла в зону воспаления. Последовательность выхода лейкоцитов получила название закона Мечникова, согласно которому спустя несколько часов с момента действия альтерирующего фактора интенсивно эмигрируют нейтрофилы, а затем моноциты и лимфоциты. Эмиграции лейкоцитов предшествует их краевое стояние у внутренней поверхности эндотелия капилляров. В механизмах краевого стояния лейкоцитов важная роль отводится замедлению кровотока в капиллярах, когда лейкоциты начинают соприкасаться с фибриновой пленкой эндотелия и удерживаться нитями фибрина. Большое значение в прилипании лейкоцитов к поверхности эндотелия имеют молекулы адгезии, экспрессируемые на поверхности лейкоцитов, люминальной поверхности эндотелия сосудов и макромолекулах межклеточного матрикса. К адгезивным молекулам относятся L-селектины, постоянно присутствующие на мембранной поверхности нейтрофилов, макрофагов и лимфоцитов, Е– и Р-селектины, появляющиеся на поверхности эндотелиоцитов после стимуляции антигеном и лимфокинами, а также интегрины, обеспечивающие как межклеточную адгезию, так и взаимодействие клеток с матриксом. Важную роль в механизмах адгезии и эмиграции лейкоцитов играет устранение отрицательного заряда эндотелиаяльной клетки и лейкоцита за счет накопления в очаге воспаления ионов водорода и калия, а также катионных белков, выделяемых лейкоцитами. Наиболее значимыми факторами инициации адгезии лейкоцитов к стенке сосудов являются комплемент, фибронектин, иммуноглобулины, гистамин, лейкотриены. После адгезии к цитомембране эндотелиальной клетки лейкоциты перемещаются через межэндотелиальные щели в подэпительнальное пространство, а оттуда проходят при участии протеиназ и катионных белков через базальную мембрану в зону альтерации. Направление движения лейкоцитов определяется хемоаттрактантами, в роли которых могут выступать продукты специфических реакций – компоненты комплемента, лимфокины, цитофильные антитела, иммунные комплексы, а также эндогенные или экзогенные неспецифические продукты метаболизма, биологически активные соединения, протеазы, эндотоксины, кинины, коллаген, плазминогенный активатор и др. В большинстве случаев острого воспаления доминирующее положение в эмиграции в течение первых 6 – 24 ч занимают нейтрофилы, через 24 – 48 ч – моноциты и несколько позднее – лимфоциты. Подобная последовательность эмиграции обусловлена, в частности, выделением нейтрофилами хемотаксических факторов для моноцитов. Однако рассмотренная последовательность эмиграции может быть иной. В частности, в зоне воспалительного процесса, индуцируемого возбудителями туберкулеза, листериоза, хламидиоза, токсоплазмоза и др., первоначально доминирует эмиграция мононуклеаров и воспаление нередко приобретает изначально хронический характер. Касаясь значимости эмигрировавших в зону воспаления лейкоцитов, следует отметить, что нейтрофилы являются активными фагоцитами, продуцентами эндопирогенов, источником вазоактивных соединений – лейкотриенов, лейкокининов, простагландинов, свободных радикалов, неферментных катионных белков с выраженной бактерицидной активностью, лизоцима, лактоферрина, а также комплекса лизосомальных гидролаз, вызывающих деструктивные процессы в зоне альтерации. Моноциты трансформируются в тканевые макрофаги, обладают выраженной фагоцитарной активностью, очищают и подготавливают зону альтерации к последующей репарации и регенерации. Подобно нейтрофилам, моноциты продуцируют эластазу, коллагеназу и другие лизосомальные ферменты, вызывающие дезинтеграцию соединительной ткани. Стимулированные моноциты продуцируют эндопирогены – ФНО, ИЛ-1, ИЛ-6, ?-интерферрон, являются источником синтеза неспецифических факторов резистентности лизоцима, комплемента, пероксидазы, активных форм кислорода. Лимфоциты в зоне воспаления продуцируют лимфокины, обеспечивают развитие специфических иммунологических механизмов защиты и развитие аллергических реакций. Все лейкоциты в зоне воспаления подвергаются жировой инфильтрации, превращаются в гнойные тельца. В процессе апоптоза нейтрофилы теряют способность секретировать лизосомальные ферменты, а макрофаги активно фагоцитируют апоптозные нейтрофилы. Экссудация – выход жидкой части крови – является одним из признаков венозной гиперемии и в то же время определяет характер тканевых изменений в зоне воспаления. Экссудация, как правило, носит двухфазный характер и включает немедленную и замедленную фазы. Немедленная фаза завершается в среднем в течение 15 – 30 мин, обусловлена контрактильными явлениями со стороны эндотелиальных клеток, преимущественно венул, под влиянием гистамина, серотонина, брадикинина, комплемента, лейкотриенов. Замедленная фаза развивается постепенно, достигает максимума через 4 – 6 ч, длится до 100 ч, связана с повреждением сосудистой стенки лейкоцитарными факторами – лизосомальными ферментами, активными метаболитами кислорода. Помимо повышения проницаемости сосудистой стенки, факторами, инициирующими экссудацию, являются: возрастание гидродинамического давления крови, снижение внутрисосудистого онкотического давления, увеличение коллоидно-осмотического давления в тканях и повышение их гидрофильности, а также активный захват эндотелиальной клеткой мельчайших капелек плазмы и перенос их за пределы сосудистого русла в воспаленную ткань (цитопемсис). В зависимости от клеточного и биохимического состава экссудат бывает серозным, фибринозным, гнойным, геморрагическим, гнилостным, смешанным. У детей в периоде новорожденности отсутствует способность рефлекторно регулировать интенсивность сосудистой реакции в зоне воспаления вследствие несформированности нейрогенного тонуса сосудов. В связи с этим, а также с незрелостью структуры лимфоидной, соединительной тканей, центральной нервной системы, обеспечивающих у взрослого быстрое формирование местных механизмов защиты, характерными особенностями воспалительного процесса у детей в этот период жизни являются преобладание альтеративно-дегенеративных изменений в тканях, быстрая генерализация инфекции и развитие септического состояния. Изменение обмена веществ в очаге воспаления. Механизмы пролиферации при воспалении Развитие альтерации, сосудистых изменений в зоне воспаления закономерно сочетается с типовыми расстройствами метаболизма. Причем, на стадии артериальной гиперемии возникает резкое увеличение интенсивности обмена веществ в связи с усилением оксигенации, трофики воспаленной ткани за счет возрастания кровотока в системе мнкроциркуляции. Однако, последовательная смена артериальной гиперемии венозной в зоне воспаления приводит к развитию явлений престаза, стаза, резкому снижению напряжения кислорода, что обуславливает подавление окислительно-восстановительных реакций, накопление промежуточных продуктов гликолиза, липолиза, протеолиза, в частности молочной, пировиноградной, жирных кислот, аминокислот и др. Избыточное накопление кислых метаболитов лежит в основе развития в зоне альтерации в начале компенсированного, а затем декомпенсированного метаболического ацидоза. Так, при остром абсцессе рН гнойного экссудата может снизиться до 5,3 – 5,0. Наряду с гипер-Н-ионией в зоне альтерации повышается онкотическое и осмотическое давление, что связано с дестабилизацией цитоплазматических мембран и избыточным поступлением ионов калия во внеклеточную среду, возрастанием уровня гидрофильных метаболитов – продуктов протеолиза, гликолиза, липолиза, а также усиленным поступлением белков из сосудистого русла в ткани в процессе экссудации. Характеризуя состояние энергетического обеспечения клеток в зоне воспаления, следует отметить, что на фазе венозной гиперемии в связи с развитием локального метаболического ацидоза возникает комплекс типовых нарушений: набухание митохондрий, разобщение процессов окислительного фосфорилирования и дыхания, снижение уровня макроэргических соединений в клетках, подавление различных энергозависимых реакций, в частности трансмембранного переноса ионов, синтеза белков и др. В условиях дефицита кислорода, прогрессирующего на фазе венозной гиперемии, увеличивается содержание АДФ, АМФ, неорганического фосфата в клетках. Избыточные концентрации АДФ в клетках зоны альтерации обеспечивают активацию ключевого фермента гликолиза – фосфофруктокиназы, дальнейшую стимуляцию процесса гликолиза, усугубление метаболического ацидоза и формирование порочного круга в развитии патологии. В условиях ацидоза возникает выраженная дестабилизация биологических мембран, в частности цитоплазматических и лизосомальных. Секреция нейтрофилами и моноцитами протеиназ, катепсинов, миелопероксидазы, катионных белков, кислых гидролаз, эластазы в зоне альтерации воздействует на межклеточный матрикс очага воспаления, приводя к его деградации. Продукты стимулированных нейтрофилов вызывают дегрануляцию тучных клеток, активируют систему комплемента, калликреин-хининовую систему, систему свертывания крови и фибринолиза. Следует отметить, что в зоне воспаления формируются и механизмы, противодействующие деградации клеток и межклеточного матрикса. Так, активированные нейтрофилы и моноциты выделяют трансформирующий фактор роста B1 (ТФР-В1), подавляющий синтез протеолитических ферментов лейкоцитами, способствующий стабилизации матрикса. Кроме того, нейтрофилы, эозинофилы и лимфоциты в зоне воспаления подвергаются апоптозу (программированная гибель клетки). При апоптозе происходят компактизация и фрагментация хроматина без разрушения биологических мембран и освобождения ферментов в окружающую среду, что исключает дальнейшее беспредельное повреждение тканей. Апоптозные лейкоциты подвергаются макрофагальному фагоцитозу и элиминируются из зоны воспаления. Пролиферация является завершающей фазой развития воспаления, обеспечивающей репаративную пролиферацию тканей на месте очага альтерации. Размножение клеточных элементов начинается по периферии очага воспаления, в то время как в центре его могут еще сохраняться явления альтерации и экссудации. Полного развития пролиферация клеточных элементов достигнет лишь после «очищения» зоны альтерации от клеточного детрита. В связи с этим следует отметить, что процессу пролиферации предшествует формирование нейтрофильного и моноцитарного барьеров, обеспечивающих процессы фагоцитоза дегенерирующих и некротизированных клеток, возбудителей инфекции. Восстановление и замещение поврежденных тканей начинается с выхода из сосудов молекул фибриногена и образования фибрина, который формирует своеобразную сетку, каркас для последующего клеточного размножения. Уже по этому каркасу распределяются быстро образующиеся фибробласты с места их активации в очаг репарации. Деление, рост и перемещение фибробластов возможно только после их связывания с фибрином или коллагеновыми волокнами. Эта связь обеспечивается особым белком – фибронектином. Размножение фибробластов начинается по периферии зоны воспаления, обеспечивая формирование фибробластического барьера. Интенсивно размножающиеся фибробласты продуцируют кислые мукополисахариды, коллагеновые волокна и другие компоненты межуточной соединительной ткани. При этом зона воспаления не только инкапсулируется, но и возникает постепенная миграция клеточных и бесклеточных компонентов соединительной ткани от периферии к центру, формирование соединительнотканного остова на месте первичной и вторичной альтерации. Факторами, стимулирующими развитие процессов пролиферации, являются цитокины (ИЛ-1, фибронектин), фактор некроза опухоли, эпидермальный, тромбоцитарный, фибробластический факторы роста, а также умеренные концентрации биологически активных веществ, ионов водорода, полиамины, антикейлоны и др. Наряду с фибробластами размножаются и другие тканевые и гематогенные клетки. Из тканевых клеток пролиферируют эндотелиальные клетки, которые формируют новые капилляры. Фибробласты вместе с вновь образованными сосудами образуют грануляционную ткань. Это, по существу, молодая соединительная ткань, богатая клетками и тонкостенными капиллярами, петли которых выступают над поверхностью ткани в виде гранул. Основными функциями грануляционной ткани являются защитная (предотвращение влияния факторов окружающей среды на очаг воспаления) и репаративная (заполнение дефекта и восстановление анатомической и функциональной полноценности поврежденных тканей). Формирование грануляционной ткани не строго обязательно. Это зависит от величины и глубины повреждения. Грануляционная ткань обычно не развивается при заживлении ушибленных кожных ранок или мелких повреждений слизистой оболочки. Грануляционная ткань постепенно превращается в волокнистую ткань, называемую рубцом. В процессе пролиферации участвуют и органоспецифические клеточные элементы органов и тканей. С точки зрения возможностей пролиферации органоспецифических клеточных элементов все органы и ткани могут быть распределены по трем группам. К первой группе могут быть отнесены органы и ткани, клеточные элементы которых обладают активной или практически неограниченной пролиферацией, достаточной для полного восполнения дефекта структуры в зоне воспаления (эпителий кожи, слизистых оболочек полости рта, дыхательных путей, желудочно-кишечного тракта и др.). Ко второй группе относятся ткани с ограниченными регенерационными способностями (сухожилия, хрящи, связки, костная ткань, периферические нервные волокна). И, наконец, к третьей группе могут быть отнесены те органы и ткани, где органоспецифические клеточные элементы неспособны к пролиферации (сердечная мышца, скелетная мышца, нервные клетки головного и спинного мозга, эмаль зубов). При локализации воспалительного процесса в указанных тканях регенерация осуществляется за счет формирования соединительнотканного рубца при одновременной гиперплазии ультраструктур клеток, сохранившихся в окружающих очаг воспаления тканях. ЛЕКЦИЯ № 7. ЛИХОРАДКА Лихорадка – типовой патологический процесс, возникающий при воздействии пирогенов на теплорегулирующий центр, характеризующийся активной временной перестройкой терморегуляции и направленный на повышение температуры внутренней среды организма вне зависимости от температуры окружающей среды. Развитие лихорадки обусловлено смещением установочной точки температурного гомеостаза на более высокий уровень под влиянием пирогенных веществ. Экзогенные пирогены инфекционного происхождения представляют собой высокомолекулярные липополисахаридные комплексы эндотоксинов, которые являются компонентом оболочек грамотрицательных микробов и выделяются при повреждении многих бактериальных клеток. Основным носителем пирогенной активности служит содержащийся в них липоид А. Высокоактивные экзопирогены практически не обладают токсическими, антигенными свойствами и видовой пирогенной специфичностью. При повторном воздействии на организм к ним формируется толерантность. Токсический эффект липополисахаридных пирогенов в организме проявляется под влиянием доз, в сотни тысяч раз превышающих минимальную пирогенную дозу. Полагают, что их пирогенные и токсические свойства обусловлены наличием различных химических группировок. К экзогенным инфекционным пирогенам относятся также термолабильные белковые вещества, выделенные из экзотоксинов гемолитического стрептококка, дифтерийных бацилл, возбудителей дизентерии, туберкулеза и паратифов. Однако, пирогенная активность их значительно ниже, чем липополисахаридных. Вирусы, риккетсии и спирохеты вызывают развитие лихорадки, несмотря на отсутствие в них экзопирогенов. Эффект инфекционных пирогенов опосредуется через образующиеся в организме эндогенные пирогены, которые являются адекватными раздражителями гипоталамического центра терморегуляции. Эндогенные пирогены представляют гетерогенную группу биологически активных веществ, обьединенных понятием цитокины: лейкоцитарный пироген (ЛП), лейкоцитактивирующий фактор интерлейкин-1 (ИЛ-1), интерлейкин-6 (ИЛ-6), фактор некроза опухолей (ФНО), ?-интерферон, макрофагальный воспалительный белок-1а, а также менее активные катионные белки и колониестимулирующие факторы (КСФ). Они образуются в очаге инфекционного, асептического или иммуноаллергического воспаления возбужденными гранулоцитами, моноцитами крови и лимфы, тканевыми макрофагами, естественными Т-лимфоцитами киллерами, В-лимфоцитами, микроглиальными и мезангиальными элементами в результате пино– и фагоцитоза экзопирогенов или поврежденных клеточных структур, иммунных комплексов и т. п. Эндопирогены могут образовываться в лейкоцитах при воздействии на них лимфокинов и «пирогенных» стероидных гормонов типа этиохоланолона и прогестерона. В отличие от экзогенных, эндогенные пирогены не вызывают развитие толерантности при повторном воздействии на организм. Для целостного понимания механизмов развития лихорадки необходимо иметь представление о структурно-функциональной организации терморегулирующего аппарата. Одна из основных функций системы теплорегуляции заключается в создании установочной точки температурного гомеостаза. Установочная температура является результатом интегрирования сигналов, поступающих от холодовых и тепловых рецепторов к специфическим термочувствительным нейронам терморегулирующего центра. Большинство их расположены в преоптической области переднего гипоталамуса. Тепло– и холодочувствительные нейроны, образующие отдел измерения («термостат»), воспринимают через соответствующие рецепторы прямые и рефлекторные температурные влияния. Медиаторами тепловых импульсов служат серотонин и норадреналин, а холодовых – ацетилхолин. Указанные термонейроны передают импульсацию о характере температурного воздействия интернейронам аппарата сравнения («установочная точка»), обладающим спонтанной импульсной активностью, которые воспринимают информацию и формируют установочную точку температурного гомеостаза. Роль медиатора в нейронах «установочной точки» выполняет ацетилхолин. Генерируемый вставочными нейронами сигнал рассогласования передается вегетативным симпатическим, парасимпатическим и соматическим нейронам, составляющим эффекторный отдел центра теплорегуляции. Медиаторами эфферентной импульсации являются норадреналин и ацетилхолин, регулирующие механизмы теплоотдачи, теплопродукции и поддержания температуры в полном соответствии с установочной точкой температурного гомеостаза. Возникающий в интернейронах сигнал сравнения необходим для осуществления обратной связи и стабилизации функции термочувствительных нейронов, обеспечивая постоянство уровня нормальной температуры и возврат к ней после понижения или повышения ее. В развитии лихорадки выделяют три стадии: I – повышения температуры; II – установления ее на более высоком уровне; III – снижения температуры до исходного значения. Первая стадия лихорадки характеризуется ограничением теплоотдачи и последующим увеличением теплопродукции. Механизмы изменения терморегуляции в этот период могут быть представлены следующим образом. При воздействии эндопирогенов в переднем гипоталамусе образуется около 20 различных «медиаторов лихорадки». Среди них наибольшее значение в повышении установочной точки температурного гомеостаза отводится простагландинам Е (ПГЕ), которые вырабатываются под влиянием ИЛ-1, ИЛ-6 и ФНО. ПГЕ активируют аденилатциклазу и ингибируют фосфодиэстеразу, что приводит к аккумуляции ц3,5-АМФ в нейронах теплорегулирующего центра. В условиях накопления ц3,5-АМФ, ионов Na и снижения концентрации ионов кальция возрастает чувствительность нейронов к холодовым и понижается чувствительность к тепловым прямым и рефлекторным влияниям, повышаются активность вставочных нейронов аппарата сравнения и установочная точка температурного гомеостаза. В результате гипоталамический центр воспринимает нормальную температуру крови, тканевой жидкости и поток афферентной импульсации от периферических термосенсоров как сигнал охлаждения, поэтому включаются механизмы, направленные на ограничение теплоотдачи, увеличение теплопродукции и температуры внутренней среды организма. При изменении возбудимости нейронов «термостата» и «установочной точки» гипоталамического центра терморегуляции импульсация от холодовых и тепловых нейронов легко воспринимается интернейронами аппарата сравнения и передается к нейронам эффекторного отдела. Генерируемый нейронами «установочной точки» сигнал рассогласования вызывает торможение парасимпатических нейронов переднего гипоталамуса и одновременное возбуждение симпатических нейронов заднего гипоталамуса. Это приводит к повышению продукции катехоламинов, спазму периферических сосудов, к уменьшению кровоснабжения кожи и теплоотдачи путем конвекции, иррадиации и потоотделения. Таким образом, увеличение температуры тела происходит прежде всего за счет ограничения тепловых потерь и накопления тепла в организме. Дополнительный прирост тепловой энергии возникает за счет несократительного и сократительного термогенеза. Несократительный термогенез обусловлен активацией механизмов химической терморегуляции. Под влиянием высоких концентраций катехоламинов в различных тканях и органах, особенно в печени, а у детей в бурой жировой ткани, возрастает потребление кислорода, стимулируется катаболизм жиров и углеводов, возникает частичное разобщение свободного дыхания и окислительного фосфорилирования. увеличиваются теплопродукция и температура тела. При недостаточном накоплении первичной теплоты и несоответствии ее установочной точке температурного гомеостаза дальнейший прирост тепловой энергии в организме осуществляется за счет включения сократительного термогенеза. В условиях ограниченного притока теплой крови к ряду внутренних органов и к кожным покровам происходит их охлаждение, возбуждение холодовых рецепторов и поступление потока афферентной импульсации к нейронам ретикулярной формации мозгового ствола, таламуса, гипоталамуса и чувствительной области коры головного мозга. В результате возникает субъективное ощущение озноба и появление соответствующих поведенческих реакций, направленных на уменьшение теплоотдачи и увеличение теплопродукции. Вследствие дальнейшей активации холодовых термонейронов переднего гипоталамуса и адренергических нейронов заднего гипоталамуса усиливаются активирующие влияния ретикулярной формации мозгового ствола на нейроны красных ядер среднего мозга и ядра черепно-мозговых нервов, на спинальные ?-, ?– и ?-мотонейроны. Это приводит к еще большей стимуляции симпатоадреналовой системы, выделению катаболических гормонов, несократительного, а затем и сократительного термогенеза путем рефлекторного повышения терморегуляторного мышечного тонуса и развития мышечной дрожи. Сократительный термогенез, обусловленный в определенной степени активацией терморецепторных нейронов в VI – VII шейных и I грудном сегментах спинного мозга, является основным источником тепла и увеличения температуры тела до уровня новой установочной точки температурного гомеостаза. Вторая стадия лихорадки заключается в том, что при повышенной теплопродукции в организме постепенно начинает возрастать теплоотдача, и эти процессы уравновешиваются. Увеличение температуры внутренней среды организма вызывает некоторую активацию тепловых рецепторов сердца, почек, вен органов брюшной полости, теплочувствительных нейронов спинного мозга и переднего гипоталамуса. Параллельно происходит ограничение импульсной активности холодовых термонейронов теплорегулирующего центра, снижение активности адренергических нейронов заднего гипоталамуса и симпатических влияний, некоторая активация парасимпатических нейронов и холинергических влияний. Все это приводит к расширению периферических сосудов, увеличению притока теплой крови к внутренним органам и коже, повышению ее температуры, потоотделения и теплоотдачи. В результате разогревания кожи и тканей понижается активность холодовых термосенсоров кожи, внутренних органов, ограничивается поток афферентной импульсации в центр терморегуляции к холодовым термонейронам переднего гипоталамуса, а от них к адренергическим нейронам заднего гипоталамуса, что сопровождается снижением симпатических влияний на периферию. В связи с этим постепенно уменьшается активирующее влияние на нейроны мезэнцефалической и бульбарной ретикулярной формации, ядер черепно-мозговых нервов и на спинальные нейроны. Одновременно снижается активность термосенситивных структур в VI – VII шейных и I грудном сегментах спинного мозга. Описанные изменения лежат в основе уменьшения сократительного и несократительного термогенеза. Усиление теплоотдачи на фоне ограничения прироста теплопродукции препятствует дальнейшему повышению температуры тела и способствует установлению ее на более высоком уровне. Третья стадия лихорадки характеризуется значительным преобладанием теплоотдачи над теплопродукцией и возвращением температуры тела к первоначальному уровню. Последнее обусловлено уменьшением концентрации пирогенов в организме, постепенным восстановлением чувствительности нейронов гипоталамического центра к холодовым и тепловым прямым и рефлекторным воздействиям. В полном соответствии с нормализацией чувствительности интернейронов аппарата сравнения установочная точка температурного гомеостаза возвращается к исходному значению. При этом происходят еще более выраженное торможение активности адренергических нейронов, снижение симпатических влияний и усиление активности парасимпатических нейронов эффекторного отдела центра терморегуляции и холинергических влияний. Параллельно отмечаются дальнейшее расширение периферических сосудов, увеличение кровоснабжения кожи, потоотделения и теплоотдачи, восстановление метаболических процессов, уменьшение теплопродукции и литическое достижение нормальной температуры тела. Установлению температуры на исходном уровне способствует возвратный поток импульсации от нейронов аппарата сравнения, стабилизирующий функцию термочувствительных нейронов аппарата измерения теплорегулирующего центра. К нормализации температуры тела приводит также восстановление импульсной активности периферических термосенсоров, расположенных в различных органах и тканях. Быстрое снижение концентрации пирогенов и прекращение их действия на гипоталамический центр могут сопровождаться развитием критического снижения температуры тела на фоне резкого расширения периферических сосудов, падения величины артериального давления и нарушения центральной гемодинамики. Лихорадка, как и любой другой типовой патологический процесс, выполняет чаще всего зашитно-приспособительную роль, но при определенных условиях может иметь патогенное значение для организма. Повышение температуры тела при ряде инфекционных заболеваний препятствует размножению многих патогенных микробов, снижает резистентность их к лекарственным препаратам. При лихорадке стимулируются метаболизм, фагоцитарная способность различных клеточных элементов, выработка антител, синтез пропердина, интерферона, активность гипоталамо-гипофизарно-надпочечниковой системы, происходит усиление выделения гормонов адаптации, возрастание барьерной и антитоксической функции печени, активируется в целом иммунобиологическая защита организма. Однако гиперпиретическая лихорадка в организме больных характеризуется преобладанием реакций повреждения и дезадаптации. Так, при индивидуальной чувствительности организма к высокой температуре, нередко возникают потеря сознания, судорожный синдром, выраженная тахикардия, гипертония и гипертензия. В условиях повышенной нагрузки объемом и сопротивлением на миокард может возникать сердечная недостаточность. Критическое падение температуры сочетается с развитием острой сосудистой недостаточности – коллапса. У детей лихорадка развивается обычно после 3 месяцев жизни. При этом температура тела повышается медленно и, как правило, не удерживается на высоком уровне, особенно на фоне колебания температуры окружающей среды. Резкий подъем температуры тела нередко не сопровождается развитием озноба и мышечной дрожи. Основным источником тепла являются у них активация метаболизма и распад бурой жировой ткани. Особенности развития лихорадки объясняются тем, что у детей первого года жизни имеют место функциональная неполноценность регуляторного центра нейрогексного сосудистого тонуса, термосенситивного рецепторного аппарата и низкая чувствительность гипоталамических нейронов к пирогенам. Кроме того, отмечаются неустойчивость обмена веществ, недостаточное потоотделение, слабое развитие скелетных мышц, теплоизолирующих свойств кожи и подкожной клетчатки, большая удельная температура тела, что выражается в несовершенстве химической и, особенно, физической терморегуляции. В условиях неполноценной физической терморегуляции не происходит существенного ограничения теплоотдачи, поэтому у значительной части детей первого года жизни лихорадка вообще не возникает. Однако при тяжелых инфекционных заболеваниях нередко отмечается высокая температурная реакция. В этих случаях повышение температуры тела связано с усилением теплопродукции в основном за счет воздействия токсических веществ, вызывающих разобщение свободного дыхания и окислительного фосфорилирования в тканях. У детей раннего возраста лихорадка может осложняться нарушением теплообмена и развитием гипертермии. У детей в возрасте старше 1 года лихорадка в неосложненных случаях развивается так же, как у взрослых. На фоне лихорадки развиваются ацидоз, значительное ограничение секреции слюны – гипосиалия, что может приводить к развитию ксеростомии и нарушению функционального состояния челюстно-лицевого аппарата. ЛЕКЦИЯ № 8. АЛЛЕРГИЧЕСКИЕ РЕАКЦИИ НЕМЕДЛЕННОГО ТИПА Аллергия (греч. «allos» – другой, иной, «ergon» – действие) – это типовой иммунопатологический процесс, возникающий на фоне воздействия антигена-аллергена на организм с качественно измененной иммунологической реактивностью и сопровождающийся развитием гиперергических реакций и повреждением тканей. Различают аллергические реакции немедленного и замедленного типа (соответственно – гуморальные и клеточные реакции). За развитие аллергических реакций гуморального типа ответственны аллергические антитела. Для проявления клинической картины аллергической реакции необходимо по крайней мере 2 контакта организма с антигеном-аллергеном. Первая доза воздействия аллергена (малая) носит название сенсибилизирующей. Вторая доза воздействия – большая (разрешающая) сопровождается развитием клинических проявлений аллергической реакции. Аллергические реакции немедленного типа могут возникать уже через несколько секунд или минут либо спустя 5 – 6 ч после повторного контакта сенсибилизированного организма с аллергеном. В ряде случаев возможна длительная персистенция аллергена в организме и, в связи с этим, практически невозможно провести четкую грань между воздействием первой сенсибилизирующей и повторной разрешающей доз аллергена. Классификация аллергических реакций немедленного типа: 1) анафилактические (атопические); 2) цитотоксические; 3) иммунокомплексная патология. Стадии аллергических реакций: I – иммунологическая II – патохимическая III – патофизиологическая. Аллергены, индуцирующие развитие аллергических реакций гуморального типа Антигены-аллергены подразделяются на антигены бактериальной и небактериальной природы. Среди небактериальных аллергенов выделяют: 1) промышленные; 2) бытовые; 3) лекарственные; 4) пищевые; 5) растительные; 6) животного происхождения. Выделяют полные антигены (детерминантные группировки + белок-носитель), способные стимулировать выработку антител и взаимодействовать с ними, а также неполные антигены, или гаптены, состоящие только из детерминантных группировок и не индуцирующие выработку антител, но взаимодействующие с готовыми антителами. Существует категория гетерогенных антигенов, имеющих сходство структуры детерминантных групп. Аллергены могут быть сильными и слабыми. Сильные аллергены стимулируют выработку большого количества иммунных или аллергических антител. В роли сильных аллергенов выступают растворимые антигены, как правило, белковой природы. Антиген белковой природы тем сильнее, чем выше его молекулярная масса и жестче структура молекулы. Слабыми являются корпускулярные, нерастворимые антигены, бактериальные клетки, антигены поврежденных клеток собственного организма. Различают также тимусзависимые аллергены и тимуснезависимые. Тимусзависимые – это антигены, которые индуцируют иммунный ответ только при условии обязательного участия 3 клеток: макрофага, Т-лимфоцита и В-лимфоцита. Тимуснезависимые антигены могут индуцировать иммунный ответ без участия Т-лимфоцитов-хелперов. Общие закономерности развития иммунологической фазы аллергических реакций немедленного типа Иммунологическая стадия начинается с воздействия сенсибилизирующей дозы аллергена и латентного периода сенсибилизации, а также включает в себя взаимодействие разрешающей дозы аллергена с аллергическими антителами. Сущность латентного периода сенсибилизации заключается, прежде всего, в макрофагальной реакции, которая начинается с узнавания и поглощения макрофагом (А-клеткой) аллергена. В процессе фагоцитоза происходит разрушение большей части аллергена под влиянием гидролитических ферментов; негидролизованная часть аллергена (детерминантные группировки) экспонируется на наружную мембрану А-клетки в комплексе с Ia-белками и и-РНК макрофага. Образовавшийся комплекс носит название суперантигена и обладает иммуногенностью и аллергогенностью (способностью индуцировать развитие иммунных и аллергических реакций), во много раз превышающей таковую первоначального нативного аллергена. В латентный период сенсибилизации вслед за макрофагальной реакцией возникает процесс специфической и неспецифической кооперации трех типов иммунокомпетентных клеток: А-клеток, Т-лимфоцитов-хелперов и антигенреагирующих клонов В-лимфоцитов. Сначала происходит распознавание аллергена и Ia-белков макрофага специфическими рецепторами Т-лимфоцитов-хелперов, затем макрофаг секретирует интерлейкин-1, стимулирующий пролиферацию Т-хелперов, которые, в свою очередь, выделяют индуктор иммуногенеза, стимулирующий пролиферацию антигенчувствительных клонов В-лимфоцитов, их дифференцировку и трансформацию в плазматические клетки – продуценты специфических аллергических антител. На процесс антителообразования влияет еще один тип иммуноцитов – Т-супрессоры, действие которых противоположно действию Т-хелперов: они тормозят пролиферацию В-лимфоцитов и превращение их в плазмоциты. В норме отношение Т-хелперов к Т-супрессорам составляет 1,4 – 2,4. Аллергические антитела подразделяются на: 1) антитела-агрессоры; 2) антитела-свидетели; 3) блокирующие антитела. Каждому типу аллергических реакций (анафилактические, цитолитические, иммунокомплексная патология) свойственны определенные антитела-агрессоры, отличающиеся иммунологическими, биохимическими и физическими свойствами. При проникновении разрешающей дозы антигена (или в случае персистенции антигена в организме) происходит взаимодействие активных центров антител с детерминантными группировками антигенов на клеточном уровне или в системном кровотоке. Патохимическая стадия заключается в образовании и освобождении в окружающую среду в высокоактивной форме медиаторов аллергии, что происходит в процессе взаимодействия антигена с аллергическими антителами на клеточном уровне или фиксации иммунных комплексов на клетках-мишенях. Патофизиологическая стадия характеризуется развитием биологических эффектов медиаторов аллергии немедленного типа и клинических проявлений аллергических реакций. Анафилактические (атонические) реакции Различают генерализованные (анафилактический шок) и местные анафилактические реакции (атопическая бронхиальная астма, аллергические ринит и конъюнктивит, крапивница, отек Квинке). Аллергены, наиболее часто индуцирующие развитие анафилактического шока: 1) аллергены антитоксических сывороток, аллогенных препаратов ?-глобулинов и белков плазмы крови; 2) аллергены гормонов белковой и полипептидной природы (АКТГ, инсулина и др.); 3) лекарственные препараты (антибиотики, в частности пенициллин, мышечные релаксанты, анестетики, витамины и др.); 4) рентгеноконтрастные вещества; 5) инсектные аллергены. Местные анафилактические реакции могут вызываться: 1) аллергенами пыльцы растений (полинозы), спор грибов; 2) аллергенами домашней и производственной пыли, эпидермиса и шерсти животных; 3) аллергенами косметических и парфюмерных средств и др. Местные анафилактические реакции возникают при попадании аллергена в организм естественным путем и развиваются в местах входных ворот и фиксации аллергенов (слизистые конъюнктивы, носовых ходов, желудочно-кишечного тракта, кожные покровы и т. д.). Антителами-агрессорами при анафилаксии являются гомоцитотропные антитела (реагины или атопены), относящиеся к иммуноглобулинам классов Е и G4, способные фиксироваться на различных клетках. Фиксируются реагины прежде всего на базофилах и тучных клетках – клетках с высокоаффинными рецепторами, а также на клетках с низкоаффинными рецепторами (макрофагах, эозинофилах, нейтрофилах, тромбоцитах). При анафилаксии выделяют две волны выброса медиаторов аллергии: 1-я волна наступает приблизительно через 15 мин, когда медиаторы освобождаются из клеток с высокоаффинными рецепторами; 2-я волна – через 5 – 6 ч, источниками медиаторов в данном случае являются клетки-носители низкоаффинных рецепторов. Медиаторы анафилаксии и источники их образования: 1) тучные клетки и базофилы синтезируют и выделяют гистамин, серотонин, эозинофильный и нейтрофильный, хемотаксический факторы, гепарин, арилсульфатазу А, галактозидазу, химотрипсин, супероксиддисмутазу, лейкотриены, простагландины; 2) эозинофилы являются источником арилсульфатазы В, фосфолипазы D, гистаминазы, катионных белков; 3) из нейтрофилов освобождаются лейкотриены, гистаминаза, арилсульфатазы, простагландины; 4) из тромбоцитов – серотонин; 5) базофилы, лимфоциты, нейтрофилы, тромбоциты и эндотелиальные клетки являются источниками образования тромбоцитактивирующего фактора в случае активации фосфолипазы А2. Клинические симптомы анафилактических реакций обусловлены биологическим действием медиаторов аллергии. Анафилактический шок характеризуется быстрым развитием общих проявлений патологии: резкого падения артериального давления вплоть до коллаптоидного состояния, расстройств центральной нервной системы, нарушений со стороны свертывающей системы крови, спазма гладкой мускулатуры дыхательных путей, желудочно-кишечного тракта, повышения проницаемости сосудов, кожного зуда. Летальный исход может наступить в течение получаса при явлениях асфиксии, тяжелого поражения почек, печени, желудочно-кишечного тракта, сердца и других органов. Местные анафилактические реакции характеризуются повышением проницаемости сосудистой стенки и развитием отеков, появлением кожного зуда, тошноты, болей в животе вследствие спазма гладкомышечных органов, иногда рвоты, озноба. Цитотоксические реакции Разновидности: гемотрансфузионный шок, резус-несовместимость матери и плода, аутоиммунные анемии, тромбоцитопении и другие аутоиммунные заболевания, компонент реакции отторжения трансплантата. Антигеном в этих реакциях является структурный компонент мембраны клеток собственного организма либо антиген экзогенной природы (бактериальная клетка, лекарственное вещество и др.), прочно фиксирующийся на клетках и изменяющий структуру мембраны. Цитолиз клетки-мишени под воздействием разрешающей дозы антигена-аллергена обеспечивается тремя путями: 1) за счет активации комплемента – комплементопосредованная цитотоксичность; 2) за счет активации фагоцитоза клеток, покрытых антителами – антителозависимый фагоцитоз; 3) через активацию антителозависимой клеточной цитотоксичности – при участии К-клеток (нулевых, или ни Т-, ни В-лимфоцитов). Основными медиаторами комплементопосредованной цитотоксичности являются активированные фрагменты комплемента. Комплементом обозначают тесно связанную систему сывороточных ферментных белков. Иммунокомплексная патология Различают генерализованную форму иммунокомплексной патологии (сывороточная болезнь) и местные реакции типа феномена Артюса. Иммунокомплексной патологии принадлежит определенное место в механизмах развития таких видов патологии, как гломерулонефрит, ревматоидный артрит, системная красная волчанка, артерииты, эндокардиты. В образовании иммунных комплексов принимают участие в качестве антигенов лекарственные препараты (пенициллин, сульфаниламидные препараты и др.), антитоксические сыворотки, аллогенные ?-глобулины, пищевые продукты (молоко, яичные белки и др.), бактериальные и вирусные аллергены. В состав иммунных комплексов при иммунокомплексной патологии входят преципитирующие и комплементсвязывающие антитела (IgG1 – 3 и IgM). Повреждающее действие обычно оказывают растворимые комплексы средних размеров, образованные в небольшом избытке антигена. Принципы и методы гипосенсибилизации Под гипосенсибилизацией подразумевают уменьшение чувствительности к аллергену. Различают специфическую и неспецифическую гипосенсибилизацию. Специфическая гипосенсибилизация – это снятие гиперчувствительности к определенному антигену. Специфическая гипосенсибилизация может быть осуществлена за счет: 1) устранения контакта с определенным антигеном-аллергеном; 2) введения малых доз антигена по различным схемам (при этом активируется выработка блокирующих антител и Т-супрессорная функция); 3) дробного введения лечебных антитоксических сывороток по Безредко (такой способ введения сопровождается постепенной элиминацией антител из организма вследствие связывания их малым количеством поливалентных антигенов). Неспецифичеокая гипосенсибилизация – это снижение чувствительности к различным антигенам-аллергенам. В целях неспецифической гипосенсибилизации используются методы, предотвращающие развитие аллергических реакций на разных фазах. В качестве средств, подавляющих иммунологическую фазу, используются рентгеновское облучение и глюкокортикоиды. Подавление патохимической и патофизиологической фаз аллергических реакций достигается использованием комплекса фармакологических препаратов с различной направленностью действия: 1) препаратов, либо увеличивающих содержание цАМФ в клетках (?-адреномиметики ингибиторы, фосфодиэстеразы), либо уменьшающих уровень цГМФ (холинолитики), либо изменяющих их соотношение (левамизол и др.); 2) антигистаминных препаратов; 3) антагонистов серотонина (дигидроэрготамин, дигидроэрготоксин); 4) ингибиторов липоксигеназного пути обмена арахидоновой кислоты, подавляющих образование лейкотриенов; 5) антипротеазных препаратов; 6) антиоксидантов (?-токоферол и др.); 7) ингибиторов калликреин-кининовой системы; 8) противовоспалительных средств (глюкокортикоиды, салицилаты). ЛЕКЦИЯ № 9. РЕАКЦИИ ГИПЕРЧУВСТВИТЕЛЬНОСТИ ЗАМЕДЛЕННОГО ТИПА Гиперчувствительность замедленного типа (ГЗТ) – одна из форм патологии клеточного иммунитета, осуществляемого иммунокомпетентными Т-лимфоцитами против антигенов клеточных мембран. Для развития реакций ГЗТ необходима предшествующая сенсибилизация, возникающая при первичным контакте с антигеном. ГЗТ развивается у животных и людей через 6 – 72 ч после проникновения в ткани разрешающей (повторной) дозы антигена-аллергена. Виды реакции ГЗТ: 1) инфекционная аллергия; 2) контактный дерматит; 3) отторжение трансплантата; 4) аутоиммунные заболевания. Антигены-аллергены, индуцирующие развитие реакции ГЗТ: 1) инфекционные (бактерии, грибы, вирусы, простейшие паразиты); 2) клетки собственных тканей с измененной антигенной структурой (аутоантигены); 3) специфические антигены опухолей; 4) белковые антигены гистосовместимости; 5) комплексные соединения, образующиеся при взаимодействии некоторых химических веществ (мышьяк, кобальт) с белками тканей. Основными участниками реакций ГЗТ являются Т-лимфоциты (CD3). Т-лимфоциты образуются из недифференцированных стволовых клеток костного мозга, которые пролиферируют и дифференцируются в тимусе, приобретая свойства антиген-реактивных тимусзависимых лимфоцитов (Т-лимфоцитов). Эти клетки расселяются в тимусзависимые зоны лимфатических узлов, селезенки, а также присутствуют в крови, обеспечивая реакции клеточного иммунитета. Субпопуляции Т-лимфоцитов: 1) Т-эффекторы (Т-киллеры, цитотоксические лимфоциты) – разрушают опухолевые клетки, генетически чужеродные клетки трансплантатов и мутировавшие клетки собственного организма, выполняя функцию иммунологического надзора; 2) Т-продуценты лимфокинов – участвуют в реакциях ГЗТ, выделяя медиаторы ГЗТ (лимфокины); 3) Т-модификаторы (Т-хелперы (CD4), амплифайеры) – способствуют дифференцировке и пролиферации соответствующего клона Т-лимфоцитов; 4) Т-супрессоры (CD8) – ограничивают силу иммунного ответа, блокируя размножение и дифференцировку клеток Т– и В-ряда; 5) Т-клетки памяти – Т-лимфоциты, сохраняющие и передающие информацию об антигене. Общие механизмы развития реакции гиперчувствительности замедленного типа Антиген-аллерген при попадании в организм фагоцитируется макрофагом (А-клетка), в фаголизосоме которого под воздействием гидролитических ферментов происходит разрушение части антигена-аллергена (около 80 %). Нефрагментированная часть антигена-аллергена в комплексе с молекулами Ia-белка экспрессируется на мембране А-клетки в виде суперантигена и представляется антигенраспознающим Т-лимфоцитам. Вслед за макрофагальной реакцией идет процесс кооперации А-клетки и Т-хелпера, первым этапом которого является распознавание антигенспецифичными рецепторами на мембране Т-хелперов чужеродного антигена на поверхности А-клетки, а также распознавание Ia-белков макрофага специфическими рецепторами Т-хелпера. Далее А-клетки продуцируют интерлейкин-1 (ИЛ-1), стимулирующий пролиферацию Т-хелперов (Т-амплифайеров). Последние выделяют интерлейкин-2 (ИЛ-2), который активирует и поддерживает бласттрансформацию, пролиферацию и дифференцировку антигенстимулированных Т-продуцентов лимфокинов и Т-киллеров в регионарных лимфатических узлах. При взаимодействии Т-продуцентов-лимфокинов с антигеном секретируются более 60 растворимых медиаторов ГЗТ-лимфокинов, которые действуют на различные клетки в очаге аллергического воспаления. Классификация лимфокинов. I. Факторы, влияющие на лимфоциты: 1) фактор переноса Лоуренса; 2) митогенный (бластогенный) фактор; 3) фактор, стимулирующий Т– и В-лимфоциты. II. Факторы, влияющие на макрофаги: 1) миграциоингибирующий фактор (MIF); 2) фактор, активирующий макрофаги; 3) фактор, усиливающий пролиферацию макрофагов. III. Цитотоксические факторы: 1) лимфотоксин; 2) фактор, тормозящий синтез ДНК; 3) фактор, ингибирующий стволовые гемопоэтические клетки. IV. Хемотаксические факторы для: 1) макрофагов, нейтрофилов; 2) лимфоцитов; 3) эозинофилов. V. Антивирусные и антимикробные факторы – ?-интерферон (иммунный интерферон). Наряду с лимфокинами в развитии аллергического воспаления при ГЗТ играют роль и другие БАВ: лейкотриены, простагландины, лизосомальные ферменты, кейлоны. Если Т-продуценты лимфокинов реализуют свой эффект дистантно, то сенсибилизированные Т-киллеры оказывают прямое цитотоксическое действие на клетки-мишени, которое осуществляется в три стадии. I стадия – распознавания клетки-мишени. Т-киллер прикрепляется к клетке-мишени посредством клеточных рецепторов к специфическому антигену и антигенам гистосовместимости (Н-2Д и Н-2К-протеинам – продуктам генов D и К локусов МНС). При этом возникает тесный мембранный контакт Т-киллера и клетки-мишени, что приводит к активации метаболической системы Т-киллера, осуществляющей в дальнейшем лизис «клетки-мишени». II стадия – летального удара. Т-киллер оказывает непосредственное токсическое воздействие на клетку-мишень за счет активации ферментов на мембране эффекторной клетки. III стадия – осмотического лизиса клетки-мишени. Эта стадия начинается с серии последовательных изменений мембранной проницаемости клетки-мишени и завершается разрывом клеточной мембраны. Первичное повреждение мембраны приводит к быстрому поступлению в клетку ионов натрия и воды. Гибель клетки-мишени наступает в результате осмотического лизиса клетки. Фазы аллергических реакций замедленного типа: I – иммунологическая – включает период сенсибилизации после введения первой дозы антигена-аллергена, пролиферацию соответствующих клонов Т-лимфоцитов-эффекторов, распознавание и взаимодействие с мембраной клетки-мишени; II – патохимическая – фаза освобождения медиаторов ГЗТ (лимфокинов); III – патофизиологическая – проявление биологических эффектов медиаторов ГЗТ и цитотоксических Т-лимфоцитов. Отдельные формы ГЗТ Контактные дерматиты Аллергия этого типа чаще возникает к низкомолекулярным веществам органического и неорганического происхождения: различным химическим веществам, краскам, лакам, косметическим препаратам, антибиотикам, пестицидам, соединениям мышьяка, кобальта, платины, воздействующим на кожу. Контактные дерматиты могут вызывать также вещества растительного происхождения – семена хлопка, цитрусовые. Аллергены, проникая в кожу, образуют стабильные ковалентные связи с SH– и NН2-группами протеинов кожи. Эти конъюгаты обладают сенсибилизирующими свойствами. Сенсибилизация обычно возникает в результате длительного контакта с аллергеном. При контактных дерматитах патологические изменения наблюдаются в поверхностных слоях кожи. Отмечаются инфильтрация воспалительными клеточными элементами, дегенерация и отслойка эпидермиса, нарушение целостности базальной мембраны. Инфекционная аллергия ГЗТ развивается при хронических бактериальных инфекциях, вызываемых грибами и вирусами (туберкулезе, бруцеллезе, туляремии, сифилисе, бронхиальной астме, стрептококковой, стафилококковой и пневмококковой инфекциях, аспергиллезе, бластомикозе), а также при заболеваниях, вызываемых простейшими (токсоплазмоз), при глистных инвазиях. Сенсибилизация к микробным антигенам обычно развивается при воспалении. Не исключена возможность сенсибилизации организма некоторыми представителями нормальной микрофлоры (нейссерии, кишечная палочка) или патогенными микробами при их носительстве. Отторжение трансплантата При трансплантации организм реципиента распознает чужеродные трансплантационные антигены (антигены гистосовместимости) и осуществляет иммунные реакции, ведущие к отторжению трансплантата. Трансплантационные антигены есть во всех ядросодержащих клетках, за исключением клеток жировой ткани. Виды трансплантатов 1. Сингенные (изотрансплантат) – донор и реципиент являются представителями инбредных линий, идентичных в антигенном отношении (монозиготные близнецы). К категории сингенных относят аутотрансплантат при пересадке ткани (кожи) в пределах одного организма. В этом случае отторжения трансплантата не происходит. 2. Аллогенные (гомотрансплантат) – донор и реципиент являются представителями разных генетических линий внутри одного вида. 3. Ксеногенные (гетеротрансплантат) – донор и реципиент относятся к различным видам. Аллогенные и ксеногенные трансплантаты без применения иммуносупрессивной терапии отторгаются. Динамика отторжения кожного аллотрансплантата В первые 2 дня пересаженный кожный лоскут сливается с кожей реципиента. В это время устанавливается кровообращение между тканями донора и реципиента и трансплантат имеет вид нормальной кожи. На 6 – 8-й день появляются отечность, инфильтрация трансплантата лимфоидными клетками, локальный тромбоз и стаз. Трансплантат становится синюшным и твердым, происходят дегенеративные изменения в эпидермисе и волосяных фолликулах. К 10 – 12-му дню трансплантат отмирает и не регенерирует даже при пересадке к донору. При повторной пересадке трансплантата от того же донора патологические изменения развиваются быстрее – отторжение происходит на 5-й день или ранее. Механизмы отторжения трансплантата 1. Клеточные факторы. Сенсибилизированные антигенами донора лимфоциты реципиента после васкуляризации трансплантата мигрируют в трансплантат, оказывая цитотоксическое действие. В результате воздействия Т-киллеров и под влиянием лимфокинов нарушается проницаемость мембран клеток-мишеней, что приводит к освобождению лизосомальных ферментов и повреждению клеток. На более поздних стадиях в разрушении трансплантата участвуют и макрофаги, усиливающие цитопатогенное действие, вызывая разрушение клеток по типу антителозависимой клеточной цитотоксичности за счет имеющихся на их поверхности цитофильных антител. 2. Гуморальные факторы. При аллотрансплантации кожи, костного мозга, почки часто образуются гемагглютинины, гемолизины, лейкотокеины и антитела к лейкоцитам и тромбоцитам. При реакции антиген-антитело образуются биологически активные вещества, повышающие проницаемость сосудов, что облегчает миграцию Т-киллеров в пересаженную ткань. Лизис эндотелиальных клеток в сосудах трансплантата приводит к активации процессов свертывания крови. Аутоиммунные заболевания Заболевания аутоиммунной природы разделяют на две группы. Первую группу представляют коллагенозы – системные заболевания соединительной ткани, при которых в сыворотке крови обнаруживаются аутоантитела без строгой органной специфичности. Так, при СКВ и ревматоидном артрите выявляются аутоантитела к антигенам многих тканей и клеток: соединительной ткани почек, сердца, легких. Ко второй группе относят заболевания, при которых в крови обнаруживают органоспецифические антитела (тиреоидит Хашимото, пернициозная анемия, болезнь Аддисона, аутоиммунная гемолитическая анемия и т. д.). В развитии аутоиммунных заболеваний выделяют несколько возможных механизмов. 1. Образование аутоантител против естественных (первичных) антигенов – антигенов иммунологически забарьерных тканей (нервной, хрусталика, щитовидной железы, яичек, спермы). 2. Образование аутоантител против приобретенных (вторичных) антигенов, образующихся под влиянием повреждающего воздействия на органы и ткани патогенных факторов неинфекционной (тепло, холод, ионизирующее излучение) и инфекционной (микробных токсинов, вирусов, бактерий) природы. 3. Образование аутоантител против перекрестно-реагирующих или гетерогенных антигенов. Мембраны некоторых разновидностей стрептококка имеют антигенное сходство с сердечными тканевыми антигенами и антигенами базальной мембраны почечных клубочков. В связи с этим антитела к названным микроорганизмам при стрептококковых инфекциях реагируют с тканевыми антигенами сердца и почек, приводя к развитию аутоиммунного поражения. 4. Аутоиммунные поражения могут возникать в результате срыва иммунологической толерантности к собственным неизмененным тканям. Срыв иммунологической толерантности может быть обусловлен соматическими мутациями лимфоидных клеток, что приводит либо к появлению мутантных запретных клонов Т-хелперов, обеспечивающих развитие иммунного ответа на собственные неизменные антигены, либо к дефициту Т-супрессоров и, соответственно, повышению агрессивности В-системы лимфоцитов против нативных антигенов. Развитие аутоиммунных заболеваний обусловлено сложным взаимодействием аллергических реакций клеточного и гуморального типа с преобладанием той или иной реакции в зависимости от характера аутоиммунного заболевания. Принципы гипосенсибилизации При аллергических реакциях клеточного типа используют, как правило, методы неспецифической гипосенсибилизации, направленной на подавление афферентного звена, центральной фазы и эфферентного звена гиперчувствительности замедленного типа. Афферентное звено обеспечивается тканевыми макрофагами – А-клетками. Подавляют афферентную фазу синтетические соединения – циклофосфамид, азотистый иприт, препараты золота. Для подавления центральной фазы реакций клеточного типа (включающей процессы кооперации макрофагов и различных клонов лимфоцитов, а также пролиферацию и дифференцировку антигенреактивных лимфоидных клеток) используют различные иммунодепрессанты – кортикостероиды, антиметаболиты, в частности, аналоги пуринов и пиримидинов (меркаптопурин, азатиоприн), антагонисты фолиевой кислоты (аметоптерин), цитотоксические вещества (актиномицин С и D, колхицин, циклофосфамид). Для подавления эфферентного звена реакций гиперчувствительности клеточного типа, включающего повреждающее воздействие на клетки-мишени Т-киллеров, а также медиаторов аллергии замедленного типа – лимфокинов, используют противовоспалительные препараты – салицилаты, антибиотики с цитостатическим действием – актиномицин С и рубомицин, гормоны и биологические активные вещества, в частности кортикостероиды, простагландины, прогестерон, антисыворотки. Следует отметить, что большинство используемых иммунодепрессивных препаратов не вызывает селективного ингибирующего воздействия лишь на афферентную, центральную или эфферентную фазы аллергических реакций клеточного типа. Следует отметить, что в громадном большинстве случаев аллергические реакции имеют сложный патогенез, включая наряду с доминирующими механизмами реакций гиперчувствительности замедленного (клеточного) типа и вспомогательные механизмы аллергии гуморального типа. В связи с этим для подавления патохимической и патофизиологической фаз аллергических реакций целесообразно сочетание принципов гипосенсибилизации, используемых при аллергии гуморального и клеточного типов. ЛЕКЦИЯ № 10. ИММУНОДЕФИЦИТНЫЕ СОСТОЯНИЯ Иммунодефицитные состояния (ИДС) – это состояния, характеризующиеся снижением активности или неспособностью организма к эффективному осуществлению реакций клеточного и/или гуморального звена иммунитета. По происхождению все ИДС подразделяются на: 1) физиологические; 2) первичные (наследственные, врожденные); 3) вторичные (приобретенные). По преимущественному повреждению клеток иммунокомпетентной системы различают 4 группы ИДС: 1) с преимущественным повреждением клеточного иммунитета («Т-зависимые», «клеточные»); 2) с преимущественным повреждением гуморального иммунитета («В-зависимые», «гуморальные»); 3) с поражением системы фагоцитоза («А-зависимые»); 4) комбинированные, с поражением клеточного и гуморального звеньев иммунитета. Физиологическая (транзиторная) гипогаммаглобулинемия новорожденных К моменту рождения у здоровых детей в крови содержатся материнские IgG и небольшое количество собственных IgG, IgM, IgA. Иммуноглобулины, полученные от матери, содержат антитела против всех видов микробов, с которыми контактировала мать, благодаря чему ребенок оказывается защищенным против них на протяжении первых месяцев жизни. Уровень материнских иммуноглобулинов постепенно снижается. Максимально дефицит их наблюдается через 2 – 3 месяца после рождения. Затем уровень собственных иммуноглобулинов ребенка в крови начинает постепенно повышаться и количество IgM достигает нормального уровня взрослого человека в конце 1-го (мальчиков) или 2-го (девочек) года жизни, IgG – после 6 – 8 лет, IgA – после 9 – 12 и IgE – лишь спустя 10 – 15 лет. Первичные ИДС Первичные ИДС – это генетически обусловленная особенность организма реализовать то или иное звено иммунного ответа. Они обусловлены генетическим блоком на различных уровнях преобразования стволовых клеток в Т– и В-лимфоциты или на последующих этапах их дифференцировки. От уровня дефекта зависит проявление ИДС. ИДС с преимущественным нарушением клеточного звена иммунитета Синдром Ди-Джорджи – возникает при гипо– и аплазии вилочковой железы. Синтез гуморальных антител не нарушен, но отмечается дефект в дифференцировке стволовых клеток в Т-клетки. Характерны частые инфекции дыхательных и мочевыводящих путей, упорные расстройства пищеварения. Лимфоцитарная дисгенезия (синдром Незелофа) – количественная и качественная недостаточность Т-системы в результате атрофии тимуса и лимфатических узлов. Характеризуется гнойно-воспалительными очагами во внутренних органах и в коже. Дети чаще погибают в первые месяцы жизни от сепсиса. ИДС с преимущественным повреждением В-системы Болезнь Брутона – возникает при дефекте созревания предшественников В-клеток в В-лимфоциты. Болеют только мальчики. Содержание ?-глобулинов в сыворотке крови составляет менее 1 %. Резко снижена резистентность к условно-патогенным бактериям, грибам. Часто возникают воспалительные заболевания слизистых оболочек, кожных покровов и паренхиматозных органов, в то время как резистентность к вирусам не нарушена. Антигенная стимуляция не приводит к усилению синтеза антител. Содержание лимфоцитов в периферической крови соответствует норме, однако в лимфоидных органах не обнаруживаются плазматические клетки. Селективные проявления иммунодефицита Возможно развитие ИДС с селективным нарушением синтеза IgG, IgA или IgM. В основе их формирования могут лежать как блокада развития отдельных субпопуляций В-лимфоцитов, так и повышение активности супрессорных Т-лимфоцитов (что бывает чаще). У больных с селективным иммунодефицитом наблюдаются рецидивирующие инфекции слизистых оболочек верхних дыхательных путей и желудочно-кишечного тракта. Дефицит секреторных IgA в слизистых оболочках пищевого канала проявляется как рецидивирующий герпетический стоматит, хронический гастрит, кишечные инфекции. ИДС с поражением системы фагоцитоза – см. лекцию «Патология фагоцитоза». Комбинированные ИДС характеризуются нарушением дифференцировки стволовых клеток, блоком созревания Т– и В-лимфоцитов и их дефицитом. Синдром ретикулярной дисгенезии характеризуется уменьшением в костном мозге количества стволовых клеток. Характерна внутриутробная гибель плода, или дети гибнут вскоре после рождения. Швейцарский тип иммунодефицита – характеризуется поражением Т– и В-систем и, следовательно, отсутствием клеточных и гуморальных реакций иммунологической защиты. В его основе лежит дефект на уровне фермента аденозиндезаминазы, что ведет к нарушению метаболизма аденозина, блокаде синтеза гипоксантина, накоплению в тканях АТФ и в результате этого блокаде созревания Т-клеток. Проявляется на 2 – 3-м месяце жизни и характеризуется злокачественным течением. В периферической крови отмечаются лимфопения, снижеие всех классов иммуноглобулинов, возникает неспособность проявлять реакции гиперчувствительности замедленного типа. Дети редко доживают до 2-летнего возраста. Синдром Луи-Бар обусловлен дефектом созревания, снижением функции Т-лимфоцитов, уменьшением их числа в крови (особенно Т-хелперов), дефицитом иммуноглобулинов (особенно IgA, IgE, реже IgG). Наблюдаются атаксия, телеангиэктазия склер и кожи, поражение ЦНС и хронические воспалительные процессы в верхних дыхательных путях и легких, злокачественные новообразования. Синдром Вискотта – Олдрича характеризуется дефицитом периферических Т-лимфоцитов, нарушением их структуры и физико-химических свойств мембран, уменьшением клеточного иммунитета при отсутствии изменений в морфологическом строении тимуса. Продукция IgM часто снижена. Характерно снижение продукции антител к антигенам-полисахаридам, но эти больные нормально реагируют на белковые антигены. Дети страдают частыми вирусными и бактериальными инфекциями. Принципы лечения первичных ИДС Лечение зависит от типа первичной иммунологический недостаточности и включает в себя целенаправленную заместительную терапию (пересадка иммунокомпетентных тканей, трансплантация эмбрионального тимуса, костного мозга, введение готовых иммуноглобулинов – ?-глобулинов, концентрированных антител, прямое переливание крови от иммунизированных доноров, введение гормонов тимуса). Применяется активная иммунизация против частых инфекций с помощью убитых вакцин, вводятся сульфаниламиды. Вторичные ИДС Вторичные ИДС развиваются под влиянием различных экзогенных воздействий на нормально функционирующую иммунную систему. Перечень основных заболеваний, сопровождающихся вторичным иммунодефицитом, предложенный экспертами ВОЗ. 1. Инфекционные заболевания: а) протозойные и глистные болезни – малярия, токсоплазмоз, лейшманиоз, шистозоматоз и др.; б) бактериальные инфекции – лепра, туберкулез, сифилис, пневмококковые, менингококковые инфекции; в) вирусные инфекции – корь, краснуха, грипп, эпидемический паротит, ветряная оспа, острый и хронический гепатиты и др.; г) грибковые инфекции – кандидоз, кокцидиодомикоз и др. 2. Нарушения питания – истощение, кахексия, нарушения кишечного всасывания и др. 3. Экзогенные и эндогенные интоксикации – при почечной и печеночной недостаточности, при отравлении гербицидами и др. 4. Опухоли лимфоретикулярной ткани (лимфолейкоз, тимома, лимфогрануломатоз), злокачественные новообразования любой локализации. 5. Болезни обмена (сахарный диабет и др.). 6. Потери белка при кишечных заболеваниях, при нефротическом синдроме, ожоговой болезни и др. 7. Действие различных видов излучения, особенно ионизирующей радиации. 8. Сильные, длительные стрессорные воздействия. 9. Действие лекарственных препаратов (иммунодепрессанты, кортикостероиды, антибиотики, сульфаниламиды, салицилаты и др.). 10. Блокада иммунными комплексами и антителами лимфоцитов при некоторых аллергических и аутоиммунных заболеваниях. Вторичные ИДС можно разделить на 2 основные формы: 1) системные, развивающиеся вследствие системного поражения иммуногенеза (при лучевых, токсических, инфекционных, стрессорных поражениях); 2) местные, характеризующиеся регионарным поражением иммунокомпетентных клеток (локальные нарушения иммунного аппарата слизистой, кожи и других тканей, развившиеся вследствие местных воспалительных, атрофических и гипоксических нарушений). Принципы лечения вторичных ИДС 1. Заместительная терапия – использование различных иммунных препаратов (препаратов ?-глобулина, антитоксических, антигриппозных, антистафилококковых сывороток и др.). 2. Коррекция эффекторного звена. Включает воздействие на иммунную систему фармакологическими препаратами, корригирующими ее работу (декарис, диуцефон, имуран, циклофосфамид и др.), гормонами и медиаторами иммунной системы (препараты тимуса – тимозин, тималин, Т-активин, лейкоцитарные интерфероны). 3. Выведение ингибирующих факторов, связывающих антитела и блокирующих эффект иммунокоррекции (гемосорбция, плазмаферез, гемодиализ, лимфоферез и др.). Ярким примером вторичного ИДС является синдром приобретенного иммунодефицита (СПИД), или ВИЧ-инфекция Этиология СПИДа. Возбудитель СПИДа относится к ретровирусам и его обозначают как ВИЧ (вирус иммунодефицита человека) или ЛАВ (лимфоаденопатический вирус). Заболевания в Европе, Америке, Австралии и Центральной Африке вызываются вирусом ВИЧ-1, а заболевания в Западной Африке – вирусом ВИЧ-2. В организм вирус проникает с кровью, с клетками при пересадке органов и тканей, переливании крови, со спермой и слюной через поврежденную слизистую или кожу. Через 6 – 8 недель после инфицирования появляются антитела к ВИЧ. Патогенез СПИДа. Возбудитель СПИДа внедряется в клетки, имеющие рецептор Т4, к которому гликопротеиды вирусной оболочки имеют высокий аффинитет (Т-хелперы, макрофаги, клетки нейроглии, нейроны). Затем происходит освобождение от вирусной оболочки и вирусная РНК выходит из сердцевинной структуры. Под влиянием обратной транскриптазы вирусная РНК становится матрицей для синтеза двунитевой ДНК, которая попадает в ядро. Далее происходит интеграция вирусспецифической ДНК в хромосомы клетки хозяина и переход вируса в следующие клеточные генерации при каждом клеточном делении. Массовая гибель Т-хелперов происходит и в связи с взаимодействием вирусного белка на поверхности зараженных клеток. Одна зараженная клетка может присоединить к себе до 500 незараженных, именно поэтому развивается лимфопения. Кроме того, подавляется способность Т-хелперов продуцировать интерлейкин-2. Снижается количество и функциональная активность естественных клеток-киллеров. Число В-лимфоцитов, как правило, остается в пределах нормы, а функциональная их активность нередко снижается. Количество макрофагов обычно не изменяется, однако наблюдается нарушение хемотаксиса и внутриклеточного переваривания чужеродных агентов. Клетки гибнут также вследствие деятельности самой иммунной системы (выработка нейтрализующих антител к белкам ВИЧ, выработка аутоантител к Т-хелперам). Все это выводит из строя иммунную защиту в целом и лишает организм способности противостоять каким-либо инфекциям. Клинические варианты заболевания СПИДом 1. Легочный тип. Характеризуется развитием пневмонии, вызванной сопутствующей инфекцией, чаще пневмоцистами. 2. С преимущественным повреждением ЦНС по типу энцефалита или менингита. 3. Желудочно-кишечный тип. Характеризуется признаками поражения ЖКТ, в первую очередь диареей (у 90 – 95 % больных). 4. Лихорадочный тип. Характеризуется возникновением длительной лихорадки, не связанной с другими заболеваниями, сопровождающейся значительным снижением массы тела, слабостью. При всех формах течения СПИДа отмечается повышенная склонность к образованию опухолей. Лечение СПИДа. Методов эффективной терапии СПИДа не существует. Лечебные мероприятия при СПИДе: 1) блокада размножения ВИЧ (подавление репликации его нуклеиновой кислоты путем ингибирования ревертазы; супрессия процессов трансляции и «сборки» вируса); 2) подавление и профилактика инфекций и опухолевого роста; 3) восстановление иммунной компетентности организма (введение препаратов тимуса, ткани костного мозга, интерлейкина-2). Физиология и патология фагоцитоза Фагоцитоз – это разновидность клеточного иммунитета, характеризующаяся распознаванием, поглощением и перевариванием фагоцитами различных чужеродных корпускулярных объектов. Различают фагоцитоз внутрисосудистый и тканевый, завершенный и незавершенный. В зависимости от размеров фагоцитируемого объекта выделяют истинный фагоцитоз – поглощение объектов размером 0,5 – 50 мкм, ультрафагоцитоз (размер объекта < 0,01 мкм), пиноцитоз (размер объекта < 0,001 мкм). Классификация фагоцитов I. По морфологическим и функциональным особенностям: 1) микрофаги – нейтрофилы, эозинофилы, базофилы; 2) макрофаги – моноциты крови и костного мозга, тканевые макрофаги (гистиоциты, купферовские клетки, альвеолярные, перитонеальные, плевральные, микроглиальные макрофаги, макрофаги селезенки, лимфатических узлов, костного мозга, остеокласты и др.). II. По способности к активному передвижению: 1) фиксированные – купферовские клетки печени, гистициты соединительной ткани, макрофаги костного мозга, лимфоузлов, синовиальных оболочек, ЦНС и др.; 2) подвижные – макрофаги серозных полостей, воспалительных экссудатов, альвеолярные макрофаги, моноциты и др. Стадии фагоцитоза: I – приближение фагоцита к объекту фагоцитоза; II – аттракция; III – поглощение объекта фагоцитом; IV – умерщвление жизнеспособных объектов (стадия киллинга); V – переваривание нежизнеспособных объектов. Стадия приближения фагоцита к объекту фагоцитоза осуществляется за счет случайного столкновения фагоцита с чужеродным объектом в кровяном русле или направленного активного движения фагоцита к объекту фагоцитоза, которое называется положительным хемотаксисом. Для осуществления процесса хемотаксиса необходимы следующие факторы: наличие на поверхности фагоцита рецепторов к хемоаттрактантам, энергии АТФ, способности фагоцита к активному передвижению, а также достаточного количества хемоаттрактантов. В роли хемоаттрактантов могут выступать продукты специфических реакций в организме (компоненты комплемента – С3а, С5а, С567, лимфокины, цитофильные антитела, иммунные комплексы и др.), эндогенные неспецифические хемоаттрактанты, выделяющиеся из поврежденных или активированных клеток, в том числе из фагоцитов, (протеазы, протеиназы, эндотоксины, калликреин, плазминогенный активатор, IgG, коллаген, цАМФ и др.). Стадия аттракции включает опсонизацию, распознавание и прикрепление фагоцита к объекту фагоцитоза. Опсонизация – процесс адсорбции на поверхности чужеродного объекта опсонинов – веществ, являющихся молекулярными посредниками при взаимодействии фагоцитов с фагоцитируемым объектом. Опсонины облегчают распознавание и повышают интенсивность фагоцитоза. Разновидности опсонинов: 1) термолабильные опсонины (компоненты комплемента – С3в, С4в, ?– и ?-глобулины, коопсонин, С-реактивный белок, фибронектин и др.); 2) термостабильные опсонины (IgG1, IgG3, IgM, агрегированные IgA1, IgA2 и др.); 3) тафтсин – тетрапептид активного центра антител. Распознавание фагоцитом объекта фагоцитоза осуществляется за счет наличия на поверхности фагоцита специфических (для IgG1 – 2, IgA, С3в, С4в, С5а и др.) и неспецифических рецепторов (для чужеродных химических структур). Прикрепление фагоцита к объекту фагоцитоза обеспечивается взаимодействием рецепторов фагоцита с поверхностью чужеродного объекта и находящимися на ней опсонинами. Стадия поглощения – активный энергозависимый процесс, заключающийся в последовательном охвате частицы псевдоподиями со всех сторон и погружении ее в цитоплазму фагоцита вместе с окружающим участком плазматической мембраны. Результатом стадии поглощения является формирование фагосомы, содержащей чужеродную частицу. Стадия киллинга обеспечивается наличием у фагоцита факторов бактерицидности, которые выделяются в фагосому или в окружающую фагоцит среду, что может обеспечить дистантный бактерицидный эффект. Классификация бактерицидных факторов фагоцитов. I. Кислородзависимые: 1) миелопероксидаза; 2) миелопероксидазнезависимые факторы – продукты «дыхательного взрыва», возникающего при активации фагоцитов (Н2О2, супероксидный анион-радикал, гидроксильный радикал, синглетный кислород, галогены и др.). II. Кислороднезависимые: 1) лизоцим; 2) лактоферрин; 3) щелочная фосфатаза; 4) катионные белки; 5) кислая среда фагосомы (рН до 4,5). Стадия переваривания возможна только в том случае, если фагоцитируемый объект утратил жизнеспособность. Переваривание обусловлено выделением в фагосому содержимого лизосом фагоцита. Лизосомы содержат около 60 различных ферментов – гидролаз (протеазы, липазы, фосфолипазы, эластазы, коллагеназы, ДНК-азы, РНК-азы, амилазы, глюкозидазы и др.). В результате слияния лизосом и фагосом формируется фаголизосома, в которой происходит окончательная деградация компонентов чужеродного объекта. Гормонально-гуморальная регуляция процесса фагоцитоза I. Активаторы процесса фагоцитоза: 1) опсонины; 2) тироксин; 3) половые гормоны; 4) цГМФ; 5) ацетилхолин и холинергические препараты. II. Факторы, тормозящие процесс фагоцитоза: 1) лейкотоксины; 2) антифагины; 3) цАМФ; 4) глюкокортикоиды. Некоторые гормональные и гуморальные вещества оказывают двойственный эффект на активность и эффективность фагоцитарного процесса. Так, известно, что адреналин активирует АМФ-циклазу и создает условия для накопления цАМФ в клетках. Однако физиологические дозы адреналина могут повышать интенсивность фагоцитоза за счет: 1) выброса лейкоцитов из депо и развития перераспределительного лейкоцитоза; 2) усиления выработки лейкопоэтина, под влиянием которого возникает истинный лейкоцитоз; 3) активации фосфорилаз, повышения интенсивности гликолиза, что обеспечивает активацию всех энергозависимых процессов в фагоцитах. Недостаточность фагоцитоза – довольно часто встречающееся состояние наследственной и приобретенной природы, которое характеризуется снижением неспецифической резистентности организма, уменьшением интенсивности антителообразования и проявляется постоянными рецидивирующими гнойно-септическими заболеваниями. Причины патологии фагоцитоза 1. Уменьшение количества фагоцитов. 2. Структурно-функциональные изменения фагоцитов врожденного и приобретенного характера. 3. Изменения гормонально-гуморальной регуляции процесса фагоцитоза и др. Уменьшение количества фагоцитов, прежде всего нейтрофильных лейкоцитов, возникает при лейкопениях врожденного и приобретенного характера, в частности при миелотоксических, выделительных, перераспределительных и иммуноаллергических лейкопениях. Снижение фагоцитарной активности может быть обусловлено следующими структурно-функциональными изменениями фагоцитов врожденного или пробретенного характера: 1) нарушением сократительных структур фагоцита; 2) изменением структуры рецепторов, чувствительных к хемотаксическим веществам и опсонинам; 3) снижением активности ферментов, осуществляющих нормальный метаболизм фагоцитов, в частности энергетический; г) дефектами бактерицидных систем фагоцитов. Интенсивность фагоцитоза определяется не только степенью зрелости и количеством фагоцитов, но и характером воздействия хемоаттрактантов. При дефиците хемоаттрактантов нарушается направленное движение активных фагоцитов, поэтому снижается интенсивность процесса фагоцитоза. Причинами нарушения фагоцитоза в данном случае могут быть: 1) врожденная или приобретенная недостаточность различных компонентов комплемента, в частности Clr, Cl, C3 – 8; 2) врожденные или приобретенные иммунодефицитные состояния по Т– и В-системам лимфоцитов, которые сопровождаются дефицитом IgG, IgM, IgA. Одним из этиологических факторов нарушения фагоцитарного процесса является дефицит опсонизирующих факторов: 1) недостаточность системы комплемента (С3b, C4b, C5b); 2) иммунологическая недостаточность первичного или вторичного происхождения, проявляющаяся нарушением продукции иммуноглобулинов. При дефиците опсонинов затрудняется узнавание чужеродного объекта, а также прикрепление фагоцита на его поверхности. Известно, что некоторые микроорганизмы, например, возбудители анаэробной газовой гангрены, продуцируют лейкотоксины и антифагины, которые вызывают развитие отрицательного хемотаксиса и нарушение различных стадий фагоцитоза. Изменения коллоидно-осмотического (гипо– и гиперосмия) и онкотического давления в среде вызывают структурно-функциональные изменения фагоцитов и снижают интенсивность фагоцитарного процесса. Декомпенсированные сдвиги кислотно-щелочного равновесия (ацидозы и алкалозы) приводят к снижению фагоцитарной активности. Эффективность неспецифических клеточных механизмов защиты во многом зависит от гормонального статуса организма. Так, недостаточный уровень тироксина, эстрогенов обуславливает снижение активности фагоцитоза. Повышение продукции глюкокортикоидов (например, при болезни и синдроме Иценко – Кушинга), или экзогенное поступление в организм высоких доз этих гормонов тормозит гистиомоно-цитарную реакцию в очагах воспаления, а также способствует незавершенности фагоцитоза, так как препятствует формированию фаголизосом. Нейромедиаторы также оказывают влияние на активность фагоцитоза. Так, ацетилхолин и холинергические препараты, повышающие уровень внутриклеточного гуанозинмонофосфата, стимулируют фагоцитоз. |
|
||
|
Главная | Контакты | Нашёл ошибку | Прислать материал | Добавить в избранное |
||||
|
|
||||